

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union; Shore CK, Worku TL, Smith CW, et al., editors. Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities. Washington (DC): National Academies Press (US); 2024 Oct 30.

Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities.
Show detailsFDA is adapting to this evolving world by embracing both the challenges and opportunities we face. We are leveraging flexibilities in our regulatory pathways to enable breakthroughs in medical science that can be translated into medical products that improve health outcomes. We are reshaping our regulatory processes and creating a nimble workforce that adapts to new technologies, medical products, biomedical science, food science, and public health.1
Robert Califf,
Commissioner of Food and Drugs (Committee on Oversight and Accountability: U.S. House of Representatives, 2024)
In the United States, the Food and Drug Administration (FDA), has authority under the Federal Food, Drug, and Cosmetic Act (FD&C Act)2 and Public Health Service Act3 to regulate medical products and devices to ensure that they are safe and effective for their intended use. In addition to protecting the public health through regulation, the FDA mission also states that it is “responsible for advancing the public health by helping to speed innovations that make medical products more effective, safer, and more affordable and by helping the public get the accurate, science-based information they need to use medical products and foods to maintain and improve their health” (FDA, 2023p).
Chapter 1 highlighted some of the many challenges faced by drug sponsors, researchers, and patients when it comes to generating evidence to support the approval of a drug to treat rare disease and conditions. The review and approval of such products by FDA likewise requires complex judgments, often based on limited information, about what it means for a drug product to be safe and effective.
FDA has long recognized the need to apply regulatory flexibility in the review and approval of marketing authorization applications. Analyses of noncancer orphan drugs have shown that over time FDA has continued to apply flexibility in the review of certain applications for orphan drug products (Sasinowski, 2011; Sasinowski et al., 2015; Valentine and Sasinowski, 2020). FDA’s 2023 guidance for industry, Rare Diseases: Considerations for the Development of Drugs and Biological Products, reiterates the need for flexibility when it comes to applying statutory standards for drug development programs to rare disease, stating, “FDA has determined that it is appropriate to exercise the broadest flexibility in applying the statutory standards, while preserving appropriate standards of safety and effectiveness, for products that are being developed to treat severely debilitating or life-threatening rare diseases” (FDA, 2023l).
As specified in the statement of task, this report focuses on the flexibilities, authorities, and mechanisms available to regulators that are applicable to rare diseases or conditions. Where available, data are provided on the impact of these activities. For more general information on the FDA drug approval process please see FDA (2022b).
This chapter is organized into sections on the following topics: drug review and approval, designation for rare disease products, expedited regulatory programs, inclusion of pediatric populations, stakeholder engagement, rare disease programs, and transparency.
DRUG REVIEW AND APPROVAL
Before initiating a clinical trial of a drug or biological product in the United States, a sponsor generally must first submit an investigational new drug (IND) application to FDA. The proposed clinical trial is generally based on pre-clinical testing and includes plans for testing the drug in humans. FDA reviews the IND application to, among other things, ensure that the proposed clinical trials do not place trial participants at unreasonable risk of harm, and that the protocol and informed consent will be reviewed by an institutional review board (IRB) that meets FDA regulatory standards (FDA, 2014a). Although the IRB is responsible for reviewing the informed consent for all clinical trials under its jurisdiction, there are situations in which an FDA review of an informed consent document in addition to IRB review is particularly important to determine whether a clinical trial may safely proceed under 21 CFR part 312 (FDA, 2014a).
After carrying out clinical trials, FDA recommends, but does not require, that the drug sponsor meet with the agency before submitting a formal new drug application (NDA) or biologics license application (BLA); these applications include pre-clinical and clinical evidence for demonstrating the safety and effectiveness of the proposed drug or biological product. After FDA receives an NDA or BLA, the agency has 60 days to decide whether to file it for review. If FDA files the NDA or BLA, an FDA review team will evaluate the drug’s safety and effectiveness. Drug products and some biological products are assigned to the Center for Drug Evaluation and Research (CDER), while other biological products (including gene therapies) and related products, including blood, vaccines, and allergenics, are assigned to the Center for Biologics Evaluation and Research (CBER). Within CDER, applications are assigned to the Office of New Drugs (OND), and then to a specific office and division within the OND based on the therapeutic area. Within CBER, applications are assigned to one of three program offices (the Office of Therapeutic Products [OTP], Office of Vaccines Research and Review [OVRR], or Office of Blood Research and Review [OBRR]) and then to a specific office and division within the OTP, OVRR, or OBRR, as appropriate, based on the treatment modality. Typically, the relevant office and division will have already been involved in the IND process. The FDA review team evaluates the NDA or BLA and decides whether to grant marketing approval (see Figure 2-1).

FIGURE 2-1
FDA drug review process. NOTES: B-R = benefit/risk; COA = clinical outcomes assessment; COI = concept of interest; COU = concept of use; PED = patient experience data; Pre-IND = preinvestigational new drug application. SOURCE: Adapted from Biotechnology (more...)
As part of the review process, FDA may seek external input through advisory committees, which include representatives from academia, industry, and patient groups. Other external input throughout the drug development lifecycle is typically collected through established governmental programs, public comment, or through special government employees. This input may be used to inform FDA’s decision but the ultimate authority and decision making resides with FDA. While the regulatory process is the same for rare and common conditions, rare disease drug development is often dependent on a limited pool of experts, many of whom may be directly involved in drug development trials and considered to have a conflict of interest. This can make it challenging to populate advisory committees with people who have relevant expertise. Additionally, as described in Chapter 4, there are a number of considerations for data sourcing, trial design, and methodologies that are particularly relevant for rare disease marketing authorization applications.
FDA strives to be transparent when sharing relevant documents following drug approval, such as letters, reviews, labels, and patient package inserts. At the same time, FDA is required by law to review and protect certain information from being released to the public, including confidential commercial information, trade secrets, and personal privacy information. FDA has stated that commercial information “is valuable data or information which is used in a business and is of such type that it is customarily held in strict confidence or regarded as privileged” (FDA, 2018c) There is an inherent tension between the legal limitations regarding the release of what could be considered confidential commercial information and FDA’s obligation to share relevant information in the interest of public health. The Food and Drug Administration Amendments Act of 2007 requires that new molecular entities/new biological entities action packages be published on CDER’s web page within 30 days after approval (see Table 2-1). However, FDA is not required to publish complete response letters—a written response sent to sponsors to indicate that FDA’s review of a marketing authorization application has been completed and the application is not ready for approval. Complete response letters usually describe “all of the specific deficiencies that the agency has identified in an application” (FDA, 2008).
TABLE 2-1
Components of an Action Package for an NDA or BLA.
Benefit–Risk Assessment
As articulated in the 2024 National Academies report, Living with ALS, there are trade-offs between enabling patient access to new medications and the degree of uncertainty when it comes to the assessment of benefit and risks for a given drug (NASEM, 2024). There are several factors involved in the assessment of the benefits and risks of a particular drug, including the clinical context, the availability of other treatments, and the seriousness of the condition (see Box 2-1). FDA guidance for industry, Rare Diseases: Considerations for the Development of Drugs and Biological Products (FDA, 2023l), notes that “a feasible and sufficient safety assessment is a matter of scientific and regulatory judgment based on the particular challenges posed by each drug and disease, including patients’ tolerance and acceptance of risk in the setting of unmet medical need and the benefit offered by the drug.” The benefit–risk assessment also involves an evaluation of the degree of uncertainty in the identified risks and benefits. For example, a small trial may not detect certain adverse events, or it may overestimate the effect of the drug. FDA guidance Benefit–Risk Assessment for New Drug and Biological Products (FDA, 2023c) states that a higher degree of uncertainty is common in rare disease drug development due to limitations on study size and that when drugs are being developed for serious diseases with few or no approved therapies, “greater uncertainty or greater risks may be acceptable provided that the standard for substantial evidence of effectiveness has been met.”
BOX 2-1
Benefit–Risk Assessment.
In the case of serious rare diseases, FDA may thus exercise regulatory flexibility by accepting clinical trials that have smaller sample sizes. Accepting smaller sample sizes for serious rare diseases places even greater importance on maximizing the trial’s potential to provide interpretable scientific evidence about the drug’s benefits and risks in order to be respectful of patients’ willingness to participate in clinical trials. Patient contribution is optimized in clinical trials (and particularly in small sample size studies) by minimizing bias and maximizing precision with trial design features such as randomization, blinding, enrichment procedures, and adequate trial duration (FDA, 2023c).
Substantial Evidence of Effectiveness
Substantial evidence of effectiveness is defined in section 505(d) FD&C Act as:
evidence consisting of adequate and well-controlled investigations, including clinical investigations, by experts qualified by scientific training and experience to evaluate the effectiveness of the drug involved, on the basis of which it could fairly and responsibly be concluded by such experts that the drug will have the effect it purports or is represented to have under the conditions of use prescribed, recommended, or suggested in the labeling or proposed labeling thereof.4
FDA has interpreted substantial evidence of effectiveness as generally requiring at least two adequate and well-controlled clinical investigations, each of which is convincing on its own (FDA, 2019a). Requiring two adequate and well-controlled clinical trials allows for independent substantiation of study results and protects against the possibility that a chance occurrence in one study would lead to an erroneous conclusion about the drug’s effectiveness (FDA, 1998, 2019a; Freilich, 2024). However, there is regulatory flexibility available in some circumstances for the amount and type of evidence needed to meet the substantial evidence standard, as described further in the FD&C Act and FDA guidance documents and discussed below.
In 1997, Congress amended the FD&C Act to clarify that substantial evidence of effectiveness could also consist of a single adequate and well-controlled study and confirmatory evidence “if [FDA] determines, based on relevant science, that data from one adequate and well-controlled clinical investigation and confirmatory evidence (obtained prior to or after such investigation) are sufficient to establish effectiveness.”5
This clarification reflected FDA’s then-current thinking given the rapidly evolving science and practice of clinical research and drug development. In 1998, FDA released subsequent guidance6 for industry, Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products (FDA, 1998), to share the agency’s thinking on the quantity and quality of data that could be used for demonstrating effectiveness of drugs.7 In this guidance, FDA provided several illustrations of the “types of evidence that could be considered confirmatory evidence, with a specific focus on adequate and well-controlled trials of the test agent in related populations or indications, as well as a number of illustrations of a single adequate and well-controlled trial supported by convincing evidence of the drug’s mechanism of action in treating a disease or condition” (FDA, 2023f). For example, a pediatric indication might be approved based on a study on adults if the pathophysiology and drug effect are similar between the populations.
In December 2019, FDA issued draft guidance, Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products, which complemented and expanded on the 1998 guidance. The new draft guidance stated, “Although FDA’s evidentiary standard for effectiveness has not changed since 1998, the evolution of drug development and science has led to changes in the types of drug development programs submitted to the Agency. Specifically, there are more programs studying serious diseases lacking effective treatment, more programs in rare diseases, and more programs for therapies targeted at disease subsets” (FDA, 2019a). Consequently, the 2019 guidance lists several illustrative examples of the types of evidence that could be considered as confirmatory of a single adequate and well-controlled study. Those examples included: (1) data from a single adequate and well-controlled study that demonstrated the drug’s effectiveness in another closely related approved indication; (2) data providing strong mechanistic support; (3) natural history data from the disease, and (4) scientific knowledge about the effectiveness of other drugs in the same pharmacological class. A single study could be sufficient for demonstrating effectiveness if the study is a large multicenter, adequate, and well-controlled trial and “the trial has demonstrated a clinically meaningful and statistically very persuasive effect on mortality, severe or irreversible morbidity, or prevention of a disease with potentially serious outcome and confirmation of the result in a second trial would be impractical or unethical” (FDA, 2019a).8
In 2023, FDA issued a draft guidance, Demonstrating Substantial Evidence of Effectiveness with One Adequate and Well-Controlled Clinical Investigation and Confirmatory Evidence, which offers further clarification on when one study and confirmatory evidence may be sufficient for establishing effectiveness (FDA, 2023f). The guidance identifies types of confirmatory evidence that could be used to supplement one clinical investigation; FDA notes that the list is not exhaustive, and that each application is considered on a case-by-case basis. The types of confirmatory evidence listed include clinical evidence from a related indication, mechanistic or pharmacodynamic evidence, evidence from a relevant animal model, evidence from other members of the same pharmacological class, natural history evidence, real-world data, and evidence from expanded access use of an investigational drug (see Chapter 4).
FDA conducts a thorough review of an application to determine whether the submitted data constitutes substantial evidence of effectiveness. FDA examines both final summaries and data from nonclinical and clinical studies, from which it determines the safety and effectiveness of the product through its own independent analysis. The review team replicates the applicant’s analyses and, if necessary, conducts additional analyses to further inform the efficacy assessment (Bugin, n.d.).
Conclusion 2-1: While the statutory requirements for drug approval for rare diseases and conditions are the same as for non-rare diseases or conditions, FDA has long recognized the need to apply regulatory flexibility in the review and approval of marketing authorization applications.
DESIGNATION FOR RARE DISEASE PRODUCTS
As discussed in Chapter 1, FDA has the authority to grant orphan drug designation for products that are used to prevent, diagnose, or treat a rare disease or condition. Additionally, FDA can award “rare pediatric disease” designation for drug applications that meet certain criteria.9 These designation programs, which are further described below, provide incentives for sponsors to develop drugs to treat rare diseases and conditions.
Orphan Drug Designation
The orphan drug designation is an FDA incentive that began in 1983 following the enactment of the Orphan Drug Act, with the goal of stimulating the development of drugs and biological products for rare diseases by lessening the financial burdens associated with orphan drug development. A drug may qualify for orphan-drug designation (ODD) if the targeted disease or condition affects fewer than 200,000 individuals in the United States at the time of the sponsor’s request, or if it affects more than 200,000 individuals but there is no reasonable expectation that the cost of developing a drug for the condition would be recovered by sales of the drug.10 To be eligible for ODD, a drug or biological product must be for a distinct rare disease or condition. FDA determines what the distinct disease or condition is by considering such factors as the pathogenesis of the condition, course of the condition, prognosis of the condition, and resistance to treatment.11 These factors are assessed in the context of the specific drug seeking ODD.
A sponsor can receive multiple designations for the same drug if it can be shown to treat more than one distinct disease or condition. A sponsor can also receive ODD for its version of a drug that is otherwise the “same drug” as an already approved drug for the same disease or condition as long as the sponsor can present a plausible hypothesis that its drug may be clinically superior to the first drug.12
A sponsor may receive ODD for a drug designed to treat an “orphan subset”13 of individuals with a disease that affects more than 200,000 people if it can be demonstrated that the subset affects less than 200,000 people in the United States and that the remaining individuals with the disease would not be appropriate candidates due to some property of the drug.14 To be considered for ODD under this “orphan subset” provision, the demonstration that other patients would not be appropriate candidates must be based on pharmacologic or biopharmaceutical properties of the drug, or previous clinical experience with the drug.15 For example, ODD could be granted if data demonstrate that the drug is only effective for patients with a certain biomarker, or that the toxicity of the drug makes it appropriate only for patients who do not respond to standard, less toxic treatments.16 FDA has clarified that an orphan subset does not refer to a clinically distinguishable subset of persons with a particular condition; eligibility for ODD under an orphan subset must be based on a property of the drug itself, not a subset of patients.17 A product targeted at just the pediatric population may be eligible for ODD in a number of ways (FDA, 2018a). First, if the disease is rare in the overall population, any product may be eligible. Second, if the disease is common in the general population, a product may be eligible for ODD if the affected pediatric population is less than 200,000 and is a valid orphan subset, meaning that the product would not be appropriate for use in the adult population owing to some property or properties of the drug; this would be considered an orphan subset designation. Third, a product may be eligible for ODD if the disease in the pediatric population is a different disease than in the adult population and the affected pediatric population is less than 200,000.
Process
Orphan drug designation is a separate process from drug approval or licensing. Sponsors seeking ODD submit a request to FDA that includes an explanation of the rare disease or condition the drug is intended to treat along with data (e.g., clinical, in vivo, and in vitro) to support the scientific rationale for using the drug in this population.18 FDA reviews the request to assess whether the drug product meets the criteria for ODD and will send the sponsor a designation letter granting ODD, a deficiency letter that requests additional information, or a denial letter (FDA, 2023h). In 2017, according to FDA’s Orphan Drug Modernization Plan, the agency aimed to complete all new ODD reviews within 90 days of receipt (FDA, 2017b).
Benefits
ODD qualifies sponsors for incentives which include tax credits of 25 percent of qualifying clinical trial expenses, a waiver of the user fee ($4 million for fiscal year 2024) (FDA, 2024o), and potentially 7-year market exclusivity—upon approval for an indication or use within the scope of the designation (Michaeli et al., 2023).19,20 In addition, the Orphan Drug Act established the Orphan Product Grants Program to provide funding for developing products for rare diseases or conditions. The statutory authority for the Orphan Products Grants Program has been expanded to include support for “prospectively planned and designed observational studies and other analyses conducted to assist in the understanding of the natural history of a rare disease.”21
Approvals of Drugs with Orphan Drug Designation
Prior to the Orphan Drug Act, few drugs were approved by FDA for rare diseases and conditions (Asbury, 1991). During the last 40 years, over 6,300 drugs have received ODD for over 1,000 rare diseases. Of the ODDs granted by FDA, 882 resulted in at least one drug approval for 392 diseases (Fermaglich and Miller, 2023). As of 2022, between 4 and 6 percent of rare diseases had an approved drug, while between 11 and 15 percent had received an ODD (Fermaglich and Miller, 2023).
In 2010, CDER approved six new molecular entities with ODD, representing 29 percent of approved new products. In 2022, CDER approved 20 new molecular entities for orphan conditions, constituting 54 percent of all center approvals (see Figure 2-2). CBER product approvals followed a similar trend, increasing from zero ODD approvals in 2010 to five in 2022, constituting 63 percent of all center approvals (see Figure 2-3). Overall, from 2010 to 2022, the number of orphan drug approvals increased in both number and percentage of the total approved new products for both CDER and CBER, with orphan drugs making up over half of approved new products for both centers in 2021 and 2022.
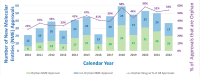
FIGURE 2-2
Proportion of CDER novel drug approvals that were orphan from 2010 to 2022. NOTE: CDER = Center for Drug Evaluation and Research. SOURCE: Presented to the Committee by Kerry Jo Lee, on November 6, 2023.

FIGURE 2-3
Proportion of CBER novel biologic approvals that were orphan from 2010 to 2022. NOTES: These data exclude in vitro diagnostic products, reagents, and intermediate biological products approved for further manufacture, such as source plasma. CBER = Center (more...)
To understand the rates of FDA approvals for non-orphan and orphan drug products over time, the committee commissioned an analysis of marketing submissions, regulatory orphan designations, and marketing approvals of new drug products from 2013–2022 using new drug applications and biologics license applications (types 1 and 1,4). Due to a lack of available data on approval and non-approval rates, the committee requested information about new drug products received from 2013–2022. Data from 2015–2020 were obtained directly from the agency for CDER and CBER. Additional data on the therapeutic areas and use of expedited review pathways were obtained from agency public assessment reports (see Appendix D for full methodology). Overall, approval rates for orphan drug product applications received between 2015 and 2020 are higher than for non-orphan drug products at nearly all FDA offices, with approval rates ranging from 83 percent (Office of Cardiology, Hematology, Endocrinology, and Nephrology) to 100 percent (Office of Immunology and Inflammation) (see Figure 2-4). Despite the increasing number of ODD and associated drug approvals, much of the progress has been concentrated in a few therapeutic areas. Between 2013 and 2022, a large portion of orphan drug approvals were for anti-cancer and immunomodulator treatments, followed by alimentary and metabolism treatments and the blood and blood forming organs treatments (see Figure 2-5).22
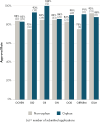
FIGURE 2-4
Novel approval rates for non-orphan and orphan new drug applications submitted to CDER from 2015 to 2020 by office. NOTES: CDER = Center for Drug Evaluation and Research; OCHEN = Office of Cardiology, Hematology, Endocrinology and Nephrology; OID = Office (more...)

FIGURE 2-5
FDA approval of orphan and non-orphan drugs by therapeutic area from 2013 to 2022. NOTES: FDA = U.S. Food and Drug Administration; NASs = new active substances; Other = other therapeutic areas not described in the top five therapeutic indications list. (more...)
Rare Pediatric Disease Priority Review Voucher Program
The rare pediatric disease priority review voucher (PRV) program, established in 2012 under section 908 of the Food and Drug Administration Safety and Innovation Act (FDASIA), was designed to incentivize the development of certain new drugs or biologics to prevent or treat rare diseases that affect pediatric populations.23 Section 908 of FDASIA defines a rare pediatric disease as “a serious or life-threatening disease in which the serious or life-threatening manifestations primarily affect individuals aged from birth to 18 years, including age groups often called neonates, infants, children, and adolescents,” and the disease is a rare disease or condition within the meaning of section 526 of the FD&C Act.24 Under this program, sponsors who receive approval for a drug to treat a rare pediatric disease may qualify for a voucher for priority review of a subsequent product (FDA, 2019d). Priority review generally means a marketing authorization submission will be reviewed by FDA in 6 months rather than the standard 10-month period review time. The sponsor may transfer or sell the voucher to another sponsor. The program has been renewed by Congress in the past, typically for 4 years at a time. Under current legislation, the rare pediatric disease designation PRV program begins to sunset after September 30, 2024 (FDA, 2024q).
Process
FDA’s draft guidance for industry Rare Pediatric Disease Priority Review Vouchers provides information on implementation of this program (FDA, 2019d). After a sponsor submits a request to FDA for rare pediatric disease designation the agency decides whether to grant the designation and whether to designate the marketing application as a “rare disease product application.” If a sponsor submits the designation request at the same time as a request for ODD or fast-track review, the “review clock” is set at 60 days.25 FDA will accept requests for rare pediatric disease designations at other times as long as requests are received prior to a filing of the NDA or BLA; these requests do not have a statutory review goal date. There are instances in which a drug may qualify for rare pediatric disease designation but not qualify for ODD and instances where a drug may qualify for ODD but not qualify for rare pediatric disease designation (FDA, 2019d). A rare pediatric disease PRV may be issued to a sponsor at the time of marketing approval if the application for the drug meets the criteria in section 529 of the FD&C Act (FDA, 2019d) and entitles the holder to priority review of an application regardless of whether the subsequent product is indicated for a rare disease.26 Sponsors also have the option of requesting a PRV independently of submitting a designation request for a rare pediatric disease.
Benefits
Drug development is a costly and time-consuming process, including the time it takes for a drug marketing authorization submission to be reviewed by regulators. A PRV helps reduce the review time for a new drug application, which can help a sponsor bring a product to market sooner. PRVs can be used by the sponsor of the original product or sold to another party; purchase prices have ranged from $67.5 million to $350 million (GAO, 2020).
The first PRV for a rare pediatric disease was awarded in 2014 (Mease et al., 2024). A 2024 report by the National Organization for Rare Disorders (NORD) showed that since the rare pediatric disease PRV program was established in 2012, there have been more than 550 rare disease pediatric designations and over 50 rare disease pediatric PRVs awarded (NORD, 2024). During the first few years of the program, Hwang et al. (2019) found that after the voucher program was implemented, drugs likely to be eligible for rare pediatric disease designation had a greater likelihood of progressing from Phase 1 to Phase 2 trials than ineligible rare disease drug products. However, no association between the launch of the program and changes in the rate of new pediatric drugs starting or completing clinical testing was found (Hwang et al., 2019). According to a 2020 U.S. Government Accountability Office (GAO) study, drug sponsors indicated that PRVs were a factor in the decision-making process for drug development (GAO, 2020). A selection of researchers interviewed by GAO reported mixed views of the rare disease PRV program—some said that the program is a useful incentive, while others indicated that some sponsors received PRVs for products they would have likely developed anyway (GAO, 2020). Other stakeholders have also shared mixed views on the program, with some saying that PRVs have been important for small companies and others saying that PRVs are a source of additional revenue for companies that do not need the money to finance drug development (GAO, 2020). Since the last GAO report, the number of rare pediatric disease PRVs granted by FDA has more than doubled, suggesting an updated assessment of the program would be helpful (NORD, 2024).
Approvals of drugs with rare pediatric disease designation
Similar to ODD, rare pediatric disease designations are concentrated for a small number of diseases. Mease et al. (2024) found that of the 245 diseases that were granted rare pediatric disease designation between 2013 and 2022, 26 diseases accounted for 41 percent of all designations.
EXPEDITED REGULATORY PROGRAMS
FDA has four general expedited programs to facilitate and expedite the development and review of certain new drugs and biological products: (1) accelerated approval, (2) fast track, (3) breakthrough therapy, and (4) priority review (see Figure 2-6). Additionally, CBER has a designation available for biologics—regenerative medicine advanced therapy (RMAT)—and FDA’s Oncology Center of Excellence has programs to expedite the review of medical products for oncologic indications. These programs are intended to facilitate and expedite the development and review of drugs that treat serious or life-threatening conditions (FDA, 2014b). FDA guidance for industry on expedited programs for serious conditions describes the eligibility criteria for expedited development and review and applies to both drugs and biologics under CDER and CBER (FDA, 2014b). Each program has its own criteria, timeline for request, and benefits, which are described in more depth below.
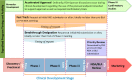
FIGURE 2-6
Entry points of expedited approval programs by clinical development stage. NOTES: BLA = biologics license application; EOP2 = end of Phase 2; IND = investigational new drug; NDA = new drug application. SOURCES: Presented to the Committee by Miranda Raggio, (more...)
Expedited regulatory pathways may be used alone or in combination with ODD or with each other; the use of these programs, particularly in combination with ODD, has increased in recent years (see Figure 2-7). To be eligible for these expedited programs, a drug must be intended to treat a serious condition and generally must represent an improvement over existing therapies (e.g., must meet an “unmet need,” show a “meaningful advantage” over existing therapies, or provide “a significant improvement in safety or effectiveness”). Due to the seriousness of and the lack of available therapies for many rare diseases, products developed to treat rare diseases may be eligible for one or more of the expedited programs offered by FDA (FDA, 2014b).
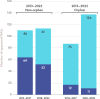
FIGURE 2-7
FDA approval rates for orphan and non-orphan drugs using expedited approval pathways from 2013 to 2022. NOTES: FDA = U.S. Food and Drug Administration; NASs = new active substances. SOURCE: CIRS Data Analysis, 2024.
Accelerated Approval
The accelerated approval pathway was established by FDA in 1992 in response to the acquired immunodeficiency syndrome (AIDS) epidemic with the goal of bringing forward new therapies for patients who desperately needed treatment options. Today, the pathway is available for drugs that meet all of the following criteria (FDA, 2014b):
- The drug “treats a serious condition”; and
- The drug “generally provides a meaningful advantage over available therapies”; and
- The drug “demonstrates an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit (i.e., an intermediate clinical endpoint).”
The pathway allows the use of a surrogate endpoint (e.g., a biomarker) or an intermediate clinical endpoint for accelerated approval—an approach viewed by some critics as subjecting patients to unnecessary risks (Kesselheim and Darrow, 2015; Redberg, 2015). Confirmatory trials are also required to verify and describe the clinical benefit (FDA, 2014b).27 In the early years of the accelerated approval program, the pathway was primarily used for drugs to treat human immunodeficiency virus (HIV) and cancer. Between 2013 and 2022, 86 percent of all drugs approved through this pathway were orphan designated drug products (see Figure 2-8). The vast majority of drugs approved through this pathway between 2010 and 2020 have been for oncology indications; 85 percent of accelerated approval drugs treated cancers (Temkin and Trinh, 2021). Although the accelerated approval pathway is not used as frequently for non-oncology rare diseases as it is for rare types of cancers, if applied appropriately, it can be a beneficial tool for bringing safe and effective drugs to patients who suffer from serious and life-threatening conditions and for whom there are no meaningful alternative treatment options (Temkin and Trinh, 2021).
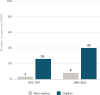
FIGURE 2-8
Number of orphan and non-orphan drug products that received accelerated approval designation by FDA between 2013 and 2022. NOTES: FDA = U.S. Food and Drug Administration; NASs = new active substances. SOURCE: CIRS Data Analysis, 2024.
As laid out in Chapter 1, the heterogeneity of rare diseases and conditions makes it difficult to identify and validate biomarkers for regulatory decision-making that are pertinent across the entire study population (Murray et al., 2023). To help fill this gap, efforts such as the Critical Path Institute’s Biomarker Data Repository, seek to advance ongoing research to advance new biomarkers for rare diseases and conditions (Critical Path Institute, n.d.).28 In 2022, Congress made several reforms to the accelerated approval program through the Food and Drug Omnibus Reform Act of 2022 to help promote transparency and more accountable use of the pathway (see Box 2-2).
BOX 2-2
Reforms and Future Direction of Accelerated Approval.
Fast Track
FDA offers fast track designation for drugs that meet one of the following criteria:
- Intended to treat a serious or life-threatening disease or condition, and nonclinical or clinical evidence demonstrate the potential to address unmet medical need;29 or
- Designated as a qualified infectious disease product30 (section 505E of the FD&C Act).
Impact
Many products developed for rare diseases meet the criteria for fast track designation—targeted at a serious or life-threatening condition and having the potential to fill an unmet medical need—at a higher rate than products developed for non-rare diseases. Using the fast track program may benefit sponsors of rare disease products by facilitating earlier and more frequent communication so that unique study issues can be discussed, and problems can be identified and addressed early in the process. Since the launch of fast track designation, it has been used primarily by orphan products, which accounted for over 50 percent of all designated products between 1998 and 2014 (Miller and Lanthier, 2016) and 64 percent of all designated products between 2013 and 2022 (see Figure 2-9).
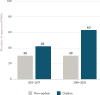
FIGURE 2-9
Number of orphan and non-orphan drug products that received fast track designation approved by FDA between 2013 and 2022. NOTES: FDA = U.S. Food and Drug Administration; NASs = new active substances. SOURCE: CIRS Data Analysis, 2024.
Breakthrough Therapy
Breakthrough therapy designation is available for drugs that meet both of the following criteria:
- Intended to treat a serious or life-threatening disease or condition; and
- Preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint(s) over available therapies (FDA, 2014b).
FDA guidance for industry, Expedited Programs for Serious Conditions (FDA, 2014b), goes into detail about the criteria that must be met for breakthrough therapy designation. FDA explains that “preliminary clinical evidence” means evidence generally from Phase 1 or Phase 2 clinical trials that is sufficient to indicate that the drug substantially improves upon available therapies but in most cases is not sufficient to establish safety and effectiveness for purposes of approval (FDA, 2014b). In general, to demonstrate a “substantial improvement,” preliminary clinical evidence should show a “clear advantage over available therapy.” The determination of whether there is substantial improvement over available therapy is a matter of judgment and depends on the size of the treatment effect and the importance of the effect to the treatment of the condition (FDA, 2014b). The “clinically significant endpoint” could be a measure of irreversible morbidity or mortality, specific symptoms that represent serious consequences of the disease, or an effect on a biomarker that strongly suggests an impact on a serious aspect of the disease (FDA, 2014b).
Due to the serious nature of rare diseases and the lack of available or adequate therapies, products aimed at rare diseases have received breakthrough therapy designation at a higher rate than products aimed at common diseases (see Table 2-2 for data on non-oncology drugs and biologics). Between 2013 and 2022, orphan medical products accounted for 75 percent of all drug products that received the breakthrough therapy designation (see Figure 2-10). Moreover, between 2018 and 2022, over half (52 percent) of novel approved orphan drugs received the designation. Experience to date has shown that the breakthrough therapy designation can dramatically speed the development and approval for selected products, improving access to effective drugs for patients with difficult-to-treat conditions (Collins et al., 2023; Shea et al., 2016).
TABLE 2-2
CDER Decisions to Grant or Deny Breakthrough Therapy Designation Requests for Non-Oncology Drugs and Biological Products from 2017 to 2019.
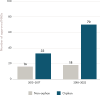
FIGURE 2-10
Number of orphan and non-orphan drug products that received breakthrough therapy designation approved by FDA between 2013 and 2022. NOTES: FDA = U.S. Food and Drug Administration; NASs = new active substances. SOURCE: CIRS Data Analysis, 2024.
Priority Review
An application for a drug can receive a priority review designation if it meets one of the following criteria:
- It is an application (original or efficacy supplement) for a drug that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness; or
- It is a supplement that proposes a labeling change pursuant to a report on a pediatric study under 505A; or
- It is an application for a drug that has been designated as a qualified infectious disease product; or
- It is an application or supplement for a drug submitted with a priority review voucher (see above for information about priority review vouchers).
“Significant improvement in safety or effectiveness” can be demonstrated in several ways, including with evidence of increased effectiveness of treatment, prevention, or diagnosis; substantial reduction of a treatment-limiting adverse drug reaction; improved patient compliance that is expected to lead to improvement in serious outcomes; or evidence of safety and effectiveness in a new subpopulation (FDA, 2014b).
Between 2008 and 2021, 62.4 percent of all drugs receiving priority review designation were orphan drugs (Monge et al., 2022). Between 2013 and 2022, 65 percent of the drug products approved through priority review received orphan designation (see Figure 2-11). Priority review can help treatments reach market more quickly, which can be critical in rare disease where there may not be another treatment option.

FIGURE 2-11
Number of orphan and non-orphan drug products that received priority review designation approved by FDA between 2013 and 2022. NOTES: FDA = U.S. Food and Drug Administration; NASs = new active substances. SOURCE: CIRS Data Analysis, 2024.
Regenerative Medicine Advanced Therapy
In addition to the programs discussed above, there is one expedited program available only for specific biologics. The 21st Century Cures Act included a provision to establish the RMAT designation to enable a new expedited option for products that meet the following criteria:
- “The drug is a regenerative medicine therapy, which is defined as a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies or products, except for those that are regulated solely under Section 361 of the Public Health Service Act and part 1271 of Title 21, Code of Federal Regulations;31
- “The drug is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and
- “Preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such disease or condition.” (FDA, 2023n)
RMAT designation is distinct from fast track and breakthrough therapy designation and has different requirements, but it includes the same benefits, such as early interactions with FDA to obtain advice on product development. In addition, there is potential for accelerated approval with post-approval requirements (FDA, 2019b). And during an open session of the committee, FDA staff reported that, as of December 31, 2022, of the 82 biologics that had received RMAT designation, 35 (43 percent) have been for orphan products (Raggio, 2023).
Oncology Center of Excellence Programs
FDA’s Oncology Center of Excellence (OCE) was established in 2017 following its authorization by the 21st Century Cures Act of 2016. The OCE brings together experts from across the agency to conduct expedited review of medical products for oncologic indications (FDA, 2024l). Between 2013 and 2022, the majority of orphan designation applications approved by FDA were for anti-cancer and immunomodulator treatments (see Figure 2-5). During this time, FDA approved 2.5 times as many orphan applications for anti-cancer and immunomodulating treatments than for nonorphan treatments. Most of the orphan approvals during this time received an expedited review.
Project Orbis
FDA’s OCE launched an international collaborative review program called Project Orbis in 2019. Applicants submitting marketing applications for products with great potential to address unmet medical needs can be considered for eligibility. Project Orbis partners can opt in or out of participating in the review of each application. The process allows for collaborative exchange of information and regulatory perspectives during the review process, while each country retains independent decision making. As of October 31, 2023, there were 81 Project Orbis applications that have resulted in product approval, with approximately one-third of these being new molecular entities in the United States (Donoghue, 2024).32 Project Orbis partners include representatives from Australia, Brazil, Canada, Israel, Singapore, Switzerland, and the United Kingdom. As of late 2023, the European Medicines Agency (EMA) and Pharmaceuticals and Medical Devices Agency (Japan’s regulatory body) became observers of the program but are not full partners in Project Orbis.
Applications in Project Orbis can be identified by the FDA review team or a sponsor can request inclusion (FDA, 2022h). Applications identified by FDA are recommended based on a combination of “breakthrough designation, impressive results, and unmet need” (FDA, 2022h). When a sponsor requests inclusion, the application is considered based on various criteria, and “high impact, clinically significant applications, should generally qualify for priority review because of improvement in safety/efficacy” (FDA, 2022h). The project is open to NDAs, BLAs, and supplemental applications for oncology indications. Sponsors are encouraged to submit marketing applications to all Project Orbis partners but are not required to do so. While review is collaborative and agencies share analysis with partners, sponsors must work with each regulatory agency to comply with regulatory requirements and timelines.
As an example of how Project Orbis works, its first review in 2019 involved FDA, Health Canada, and the Australian Therapeutic Goods Administration. The three agencies collaboratively reviewed the application and identified all regulatory divergence; all three countries simultaneously approved the drug under their own accelerated approval programs. Each country used its own format for the drug label, although they exchanged labels to note any differences (FDA, 2022h). As a result of the collaborative process, the products—for a specific type of endometrial carcinoma—were approved. The approval came 3 months prior to the FDA goal date (FDA, 2019c). For rare disease patients and their families, gaining access to new treatments even a few months earlier can mean significantly improved health outcomes, reduced suffering, and a better quality of life.
The overarching intent of Project Orbis is to leverage a global patient population and evolve toward uniform global standards (“global regulatory convergence”) for the evaluation of oncology-related orphan drugs and biologics. This is broadly applicable and appropriate for other rare diseases and aligns with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). The framework of Project Orbis that informs the concurrent submission and collaborative review process will also help the community of potential Collaboration on Gene Therapies Global Pilot (CoGenT Global) stakeholders engage productively with the program by shaping their expectations (see Box 2-3).
BOX 2-3
Pilot Program: Collaboration on Gene Therapies Global Pilot.
Real-Time Oncology Review
The real-time oncology review (RTOR) was implemented by OCE in 2018 to “facilitate earlier submission of top-line results.” To be eligible for RTOR, a product must meet all of the following criteria (FDA, 2023m):
- Clinical evidence indicates that the drug demonstrates substantial improvement on a clinically relevant endpoint(s) over available therapies; and
- Clinical trial endpoints are easily interpreted; and
- No aspect of the submission is likely to require a longer review time.
Though focused on cancers, RTOR has benefited rare diseases. Of the 10 new molecular entity (NME) product applications accepted by the initial pilot, 8 received orphan designation (Gao et al., 2022). Additionally, 11 of the 13 approvals through RTOR between 2013 and 2022 were for orphan products (see Figure 2-12).
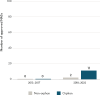
FIGURE 2-12
Number of orphan and non-orphan drug products using the real-time oncology review program approved by FDA between 2013 and 2022. NOTES: FDA = U.S. Food and Drug Administration; NASs = new active substances. SOURCE: CIRS Data Analysis, 2024.
One lesson that is relevant to rare diseases is that the program’s resource-intensive nature (e.g., submitting multiple application components at different time points as outlined in the RTOR operating procedures) means that it is key to have close coordination and alignment between the sponsors, which are often small biotech companies, and agency reviewers on expectations and submission timelines related to datasets, analysis, key claims in labels, and safety update reports among others. The RTOR program implementation has been associated with faster approval times (see Figure 2-13) although this may be confounded by the concurrent usage of other expedited programs such as breakthrough designation and priority review (Gao et al., 2022). The frequent interactions with FDA reviewers during the submission and review lifecycle due to the RTOR has meaningfully informed industry practice with respect to application preparation and FDA engagement (Kim, 2022). The abbreviated time from submission to approval means that this framework has considerable applicability to non-oncology rare diseases more broadly. Applying a similar framework may be beneficial for the advancement of rare disease drug development beyond rare cancers.
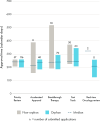
FIGURE 2-13
Approval time of orphan and non-orphan drug products approved by FDA from 2018 to 2022 by regulatory pathway. NOTES: Box plot displays median approval time with 25–75% interquartile range. Range is not displayed when n < 3. FDA = U.S. (more...)
Orphan Drugs and Expedited Review
Orphan products use expedited programs more often than non-orphan products. Figure 2-14 shows the percentage of orphan drug approvals compared with non-orphan drug approvals for NMEs and new biologics in each of the four expedited development programs available to all drugs and biologics. The number of approvals of NMEs and new biologics through each program varies; for example, in 2022, there were 30 approvals for priority review, 19 breakthrough therapy approvals, 7 accelerated approvals, and 16 fast track approvals (see Figure 2-15). Drugs and biologics may be eligible for more than one expedited program (see Figure 2-14). Median approval times for orphan and non-orphan products are generally comparable within a given expedited development pathway (see Figure 2-14). Expedited approval programs do not change FDA’s standards for approval. However, accelerated approval allows FDA to rely on a different evidence base. Surrogate endpoints or intermediate clinical endpoints can serve as the basis for accelerated approval with careful evaluation of the endpoints’ effect on clinical benefit (FDA, 2014b).

FIGURE 2-14
Use of expedited development programs in CDER and CBER from 2013 to 2022. NOTES: CBER = Center for Biologics Evaluation and Research; CDER = Center for Drug Evaluation and Research. SOURCES: Presented to the Committee by Miranda Raggio, on November 6, (more...)
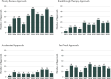
FIGURE 2-15
Expedited program approvals for new molecular entities and novel biologics in CDER and CBER from 2013 to 2022. NOTES: CBER = Center for Biologics Evaluation and Research; CDER = Center for Drug Evaluation and Research. SOURCES: Presented to the Committee (more...)
INCLUSION OF PEDIATRIC POPULATIONS
The majority of rare diseases affect the pediatric population (Wright et al., 2018). Evidence has shown there can be substantial differences in the way that children respond to drug treatment compared to adults (IOM, 2008). Thus, the inclusion of pediatric populations in clinical trials should be a core component for rare disease drug development. Epps et al. (2022) list the following challenges for rare pediatric disease drug development: “(1) garnering interest from sponsors, (2) small numbers of children affected by a particular disease, (3) difficulties with study design, (4) lack of definitive outcome measures and assessment tools, (5) the need for additional safeguards for children as a vulnerable population, and (6) logistical hurdles to completing trials, especially with the need for longer term follow-up to establish safety and efficacy.” These challenges are common across all therapeutic areas, but are amplified for rare diseases and conditions.
Over the past several decades, a combination of legislation, regulatory action (see Table 2-3 for list of guidances on pediatric drug development), and the accumulation of scientific evidence has enabled what some have considered a to be a “revolutionary change” in pediatric drug development—a shift from considering pediatric populations as “therapeutic orphans” to a current state in which the number of drug products approved for use in children continues to increase (FDA, 2024m; Fung et al., 2021).
TABLE 2-3
FDA Guidance on Pediatric Drug Development.
Two laws—the Pediatric Research Equity Act (PREA) of 2003 (FDA, 2024n), the Best Pharmaceuticals for Children Act (BPCA) of 2002 (NICHD, n.d.)—work together to address the need for pediatric drug development. Pediatric provisions in FDASIA further strengthened these laws (FDA, 2018b). BPCA provides incentives (additional marketing exclusivity) to encourage sponsors to carry out studies in pediatric populations, and PREA authorizes FDA to require pediatric studies for certain drugs and biological products (FDA, 2018b, n.d.-a; PhRMA, 2020; Sachs et al., 2012) (see Box 2-4).
BOX 2-4
Best Pharmaceuticals for Children Act (BPCA) and Pediatric Research Equity Act (PREA).
Following enactment of BPCA and PREA, FDA has reported “significant progress in the number, timeliness, and successful completion of studies in pediatric populations” (FDA, n.d.-a). The number of pediatric labeling changes—updates to a drug product’s labeling to add information on safety, effectiveness, or dosing for children—has continued to increase over time. Between 2010 and 2018, approximately one-third of the orphan drug indications approved by FDA were approved for use in children or targeted a pediatric disease (Kimmel et al., 2020). A more recent analysis showed that the percentage of orphan indications for rare pediatric diseases has continued to increase over time (Fung et al., 2021).
In 2020, PhRMA cited the following numbers:
- “Since 1998, there have been over 750 labeling changes reflecting pediatric information.
- “Since the reauthorization of BPCA and PREA in 2007, more than 680 pediatric studies have been completed under BPCA and PREA.
- “Over 250 drugs have been granted pediatric exclusivity under BPCA.
- “There are currently more than 2100 industry sponsored pediatric clinical trials underway, involving more than 1.2 million pediatric patients across a variety of therapeutic areas, including diseases where there is significant unmet need, such as infectious diseases, neurologic conditions, genetic disorders, and several forms of cancer” (PhRMA, 2020).
Notably, orphan designated drug products are generally exempted from PREA requirements. PREA states, “Unless the Secretary requires otherwise by regulation, this section does not apply to any drug for an indication for which orphan designation has been granted under section 526” (FDA, 2005). While the intent of this provision may have been to incentivize the development of drugs to treat rare diseases and conditions, patient groups, such as the Treatment Action Group and Elizabeth Glaser Pediatric AIDS Foundation, have argued that the exemption has led to the opposite result—delays or complete lack of research on the safety and efficacy of rare disease drug products for pediatric populations (Treatment Action Group and Elizabeth Glaser Pediatric AIDS Foundation, 2019).
The 2017 Research to Accelerate Cures and Equity (RACE) for Children Act33 amended PREA to remove the exemption for orphan designated drugs, but only for certain cancer drugs. A 2024 FDA briefing document to the Pediatric Oncology Subcommittee of the Oncologic Drugs Advisory Committee stated that:
“Based upon the updated FDA analysis of initial pediatric study plans for molecularly targeted drugs and the results of the GAO audit, it appears that the RACE Act has contributed to an increase in the number of planned studies to test certain molecularly targeted drugs in pediatric patients with cancer. However, given the amount of time needed to design and conduct clinical trials evaluating new drugs for the treatment of pediatric cancers, it is too early to determine the extent to which implementation of the Food and Drug Reauthorization Act of 2017 (FDARA) provisions of PREA will advance the development of new treatments for pediatric cancers” (FDA, 2024g)
Closing the Gap
Despite legislative, regulatory, and scientific advances, meta-analysis has shown that off-label prescriptions for children accounted for up to 38 percent of all pediatric prescriptions between 2007 to 2017 (Allen et al., 2018). To this day, off-label drug use remains an issue for pediatric populations, particularly those living with rare diseases and conditions for which there is no available treatment on the market (Committee on Drugs et al., 2014). More is needed to meet the needs of children living with rare diseases and conditions.
In addition to measures taken by Congress to incentivize the inclusion of pediatric populations in rare disease drug development, FDA and NIH, in partnership with nongovernmental organizations—including patient and disease advocacy groups, academic clinical investigators, and biopharmaceutical companies—have an opportunity to better collaborate on approaches to include pediatric populations as early as possible in rare disease clinical trials.
During an open session held by the committee on February 7, 2024, senior representatives from CDER, CBER, and OCE all stated the agency’s view concerning the inclusion of pediatric participants early in clinical trials is “evolving.” This sentiment has been echoed by FDA in other venues. On FDA Rare Disease Day, February 27, 2023, Martha Donoghue, the associate director for pediatric oncology and rare cancers in OCE, stated that “over the past decade, this tendency to think about protecting pediatric patients from clinical trials has shifted to thinking that we can best protect pediatric patients with cancer through more timely and thoughtful conduct of pediatric clinical trials. In essence, to give them a chance to benefit through participation in clinical trials as soon as it is scientifically justified and feasible, with the ultimate goal of providing increased access to new, safe, and effective treatment that are approved for pediatric patients with cancer” (FDA, 2023g).
Dr. Nicole Verdun, director, Office of Therapeutic Products, CBER, FDA, stated publicly on March 8, 2024, “…traditionally, the thought has been that you should try things in adults before children…what I hear from patient communities is that we have diseases, especially in the rare disease space, that affect children from birth, and we want to prevent those conditions from happening. We do not want to wait to have to enroll a young child in a trial…we have to pivot a little in mindset and be comfortable with starting sometimes in areas where there is some uncertainty… it really depends on the program, but we are looking for ways to enroll pediatric patients earlier in the development process” (The Alliance for a Stronger FDA, 2024).
Such sentiments have been reiterated in other public meetings, including a 2024 Roundtable on Effective Inclusion of Children Early in Clinical Trials hosted by Leavitt Partners and the Friedreich’s Ataxia Research Alliance, during which senior FDA leadership at the office, division, and center level discussed the need to pivot toward earlier inclusion of pediatric populations in clinical trials. The committee recognizes the need to focus on protecting children living with rare diseases and conditions through clinical trials, so they have access to safe and effective therapies.
RECOMMENDATION 2-1: Congressional action is needed to encourage and incentivize more studies that provide information about the use of rare disease drug products in pediatric populations. To that end, Congress should remove the Pediatric Research Equity Act orphan exemption and require an assessment of additional incentives needed to spur the development of drugs to treat rare diseases or conditions.34
Additionally, the U.S. Food and Drug Administration (FDA) and the National Institutes of Health in partnership with nongovernmental organizations, including patient groups, clinical investigators, and biopharmaceutical companies, should work to provide clarity regarding the evolving regulatory policies and practices for the inclusion of pediatric populations as early as possible in rare disease clinical trials. Actions should include, but are not limited to the following:
STAKEHOLDER ENGAGEMENT
As laid out in Chapter 1, rare disease drug development faces multiple challenges: lack of understanding of the underlying disease, a lack of defined endpoints, and limited patient populations.
FDA has recognized the need for its employees to supplement their knowledge, particularly for “new and emerging fields of science, pioneering technologies, and the increasing complexity of medical devices and pharmaceuticals” (FDA, 2024k). Drugs intended to treat rare diseases fall under each of these umbrellas.
FDA has a number of mechanisms in place to leverage the experience and expertise of external stakeholders, including public workshops on novel and evolving areas of development (e.g., disease-specific or method-based approaches), advisory committee meetings, and public comment on proposed regulations or guidances. Additionally, FDA can appoint special government employees, recruit qualified experts to serve on advisory committees, and collaborate with other organizations (FDA, 2016). The agency has used all of these mechanisms to support the review and approval of drugs to treat rare disease and conditions, though some of these approaches can be slow and onerous to implement.
FDASIA, signed into law in 2012, includes sections aimed at expanding the role of external stakeholders in FDA regulatory processes. Section 1137 directs the Secretary of Health and Human Services (HHS) to “develop and implement strategies to solicit the views of patients during the medical product development process and consider the perspectives of patients during regulatory discussions.”35 This provision assists FDA in developing strategies for integrating the input of patients in regulatory decision-making.
Section 1138 of FDASIA “requires the Secretary of Health and Human Services (HHS), acting through the Commissioner of Food and Drugs, review and modify as necessary, FDA’s communication plan to inform and educate health care practitioners and patients on the benefits and risks of medical products, with particular focus on underrepresented subpopulations, including racial subgroups” (FDA, 2013a). While these provisions are complementary, they serve different purposes in aiding the agency better integrate the critical input of patients and ensuring adequate communication of information about drug development.
Patient Engagement
FDA has made important strides to engage people with lived experience throughout the regulatory review process. Guidance documents, patient focused drug development (PFDD) meeting summaries, direct interaction with patients, and other resources have helped to bridge the gaps among regulators, sponsors, and people with lived experience.
An FDA guidance for industry, Rare Diseases: Considerations for the Development of Drugs and Biological Products, encourages sponsors to discuss trial design with patients and caregivers early in the planning stages, and to consider modifying designs to address patient and family concerns (FDA, 2023l). For example, the guidance notes that endpoint selection can be informed by understanding what aspects of the disease are meaningful to patients and caregivers. In addition, the guidance emphasizes the importance of centering patients in the process. Patient engagement is a critical aspect of the regulatory review process as it provides decision makers with information on lived experience, meaningful outcome measures, therapeutic context, and benefits and risks.
Patients and caregivers are experts in their own experience of living with or caring for someone with a disease or condition and their perspectives and insights should be valued alongside those of regulators, sponsors, and researchers when it comes to informing the development of drugs to meet their needs. A 2015 JAMA article by Hunter, O’Callaghan, and Califf (Hunter et al., 2015) stated that “the FDA is working to give patients a greater voice in medical product development and evaluation” (p. 2500). FDA has several avenues for patient and caregiver input to inform regulatory decision-making. These include the PFDD initiative, the FDA Patient Listening Session program, the FDA Patient Representative Program, and the FDA–Clinical Trials Transformation Initiative (CTTI) Patient Engagement Collaborative (PEC). Information shared via these avenues can provide key insights on disease course and variability, disease outcomes, patient preferences, desired benefits, and acceptable risks and has increasingly become a component of regulatory science at FDA (Kuehn, 2018).
Patient-Focused Drug Development
FDA’s PFDD initiative, which was established under the fifth authorization of the Prescription Drug User Fee Act (PDUFA), is a “systematic approach to help ensure that patients’ experiences, perspectives, needs, and priorities are meaningfully incorporated into drug development and evaluation” (FDA, 2024d) (see Box 2-5). PFDD initially included a series of FDA-hosted disease-specific meetings, which were held between 2012 and 2017, in which FDA and other key stakeholders, including medical product developers, health care providers, and federal partners, heard directly from patients, their families, caregivers, and patient advocates about the symptoms that matter most to them, the impact the disease has on patients’ daily lives, and patients’ experiences with currently available treatments. Following each meeting, FDA published a summary of the input shared by meeting participants (FDA, 2024f).
BOX 2-5
What is Patient-Focused Drug Development?
PFDD has since expanded to include externally-led PFDD meetings (FDA, 2022e), the development of guidance documents, and other initiatives to facilitate the incorporation of patient input into medical product decision making. Patient listening sessions, similar to PFDD meetings, serve to inform FDA on the concerns of the patient community. However, patient listening sessions are nonpublic, only FDA, patients, caregivers, advocates, and community representatives can participate in the sessions, and they are non-interactive in that the agency participants are listening but not conversing with other participants (FDA, 2024h). There have been over 35 rare disease-specific patient listening sessions since they began in October 2018 (FDA, 2024e) (see Appendix F for a full list of rare disease-specific PFDD meetings and patient listening sessions).
During a series of qualitative interviews with industry representatives, interviewees suggested that PFDD meetings are an adequate mechanism for FDA to gather patient input on disease-specific drug development (see Appendix E for full methodology and results). At the same time, during committee meeting open sessions, a few speakers suggested that the one-way nature of PFDD meetings and patient listening sessions makes it hard for participants to understand the value of their contributions during these convenings. The committee did not find adequate evidence to assess the impact of PFDD meetings and patient listening sessions one way or the other.
PFDD meetings and patient listening sessions provide a pathway for patient input to be considered during the review process, but patient groups often face barriers to effectively leverage these opportunities. Challenges cited in a study by Kuehn (2018) include “resources capacity, . . . policy knowledge, and regulatory culture [at FDA]” (p. 663). There is a perception that well-funded patient groups are more capable than smaller groups of generating data that meet FDA regulatory standards. Smaller advocacy groups may have limited resources to host PFDD meetings and lack full knowledge of regulatory processes and data requirements for decision making.
A broader issue raised by Kuehn (2018) was a perception on the part of interviewees that FDA did not give adequate weight to patient experience data (PED) and should put in place more practical conflict of interest policies for patient groups, which often depend on industry engagement and funding support.36
Conclusion 2-2: FDA is using available mechanisms to gather patient input. However, there are opportunities to better ensure that patient input informs the development of treatments for rare diseases as well as the design and conduct of clinical trials for rare diseases. More clarity is needed on the part of patient groups and people with lived experience on how the agency is using patient input to inform regulatory decision-making and what types of patient input are most relevant.
FDA Patient Listening Sessions
FDA patient listening sessions are small, informal meetings between FDA staff and patient communities that allow staff in various centers and review divisions the opportunity to hear patient and caregiver perspectives on a range of topics (e.g., disease/treatment, burden, impact on daily activities/quality of life, meaningful outcomes in managing their disease, perspectives on clinical trial participation) related to their experiences with a disease/condition or health care consideration for specific populations (FDA, 2024h).
FDA–CTTI Patient Engagement Collaborative
The PEC was established in 2018 as a joint project of FDA and CTTI, a public-private partnership of Duke University and FDA (CTTI, 2023). The PEC serves as a setting for patients, FDA, and CTTI to discuss patient engagement during medical product regulation. The idea for the PEC came out of public comments related to how FDA could develop strategies to solicit the views of patients (as well as a joint structure between FDA and EMA known as the Pediatric Cluster; see Chapter 5); many commenters suggested that a group outside FDA would be best suited to providing ideas about how to do so. Members of the PEC are representatives of the patient community. Meetings are held up to six times per year and may cover topics such as exploring creative ideas to enhance patient engagement, identifying needs for tools and resources for patient communities, and engaging in two-way education about medical product regulation. In 2022 the PEC held a joint meeting with EMA Patients and Consumers Working Party (see Chapter 3) to discuss common issues in patient engagement.37
Patient Representative Program
The Patient Representative Program allows for patients, caregivers, and advocates representing selected disorders chosen by FDA to serve on FDA advisory committees via appointment as a special government employee and provide advice to FDA on the regulation of medical products (FDA, 2024b).
Patient Experience Data
Patient experience data38 provide information about the experiences, perspectives, needs, and priorities of people living with a disease or condition. These data can be used to inform clinical trial design, endpoint selection, and regulatory review, including benefit–risk assessments (FDA, n.d.-b).
An analysis of NMEs approved by FDA in 2018 found that of the 59 NMEs approved, 48 contained a patient experience data table within the review documentation and 34 reported using patient experience data in the drug review process. Within this group of NMEs, 28 had received an orphan designation. However, only 17 of the 28 (60.7 percent) orphan designated NMEs used PED in the review. In comparison, 17 of the other 20 (85 percent) non-orphan reviews used PED. Sponsor-submitted patient-reported outcomes (PROs) were the most significant source of patient experience data and were used in over half of approved drug reviews. Non-PRO patient experience data used in reviews were qualitative studies, PFDD meeting summary reports, observational survey studies, natural history studies, and patient preference studies (Kieffer et al., 2019).
The 21st Century Cures Act directs FDA to report on the use of patient experience data in regulatory decision-making. A 2021 report by Eastern Research Group (ERG) showed that while patient experience data play an important role in FDA decisions about when to use PROs or other types of clinical outcome assessments, they “generally provide supporting information in situations where the condition is not well characterized, as with some rare diseases” (Eastern Research Group Inc., 2021). The report included interviews with external stakeholders, including patients and their caregivers, clinicians, and representatives of patient and disease advocacy organizations, which indicated a lack of understanding about how FDA uses patient experience data. Similar statements were made by Kara Berasi, the chief executive officer of the Haystack Project, at a committee meeting open session. The ERG report offered recommendations for FDA and other stakeholders to provide more clarity and consistency on whether and how FDA uses patient experience data in regulatory decision-making (see Box 2-6).
BOX 2-6
Select Eastern Research Group Report Findings and Recommendations.
Opportunity to Enhance Patient Input
Taken together, FDA has demonstrated a commitment to engaging people with lived experience. While there are numerous programs, collaborations, and partnerships in place that value the experience and perspectives of people living with rare diseases and conditions and their caregivers, more is needed to fully implement Section 1137 of FDASIA. This is particularly the case for patient groups that are small and under resourced. Strategies are needed to better solicit input from people no matter their race, ethnicity, ability, socio-economic level, or geography with particular consideration for enabling the participation of those who come from marginalized communities.
FDA advisory committees offer independent expert advice and recommendations on scientific, technical, and policy matters related to FDA-regulated products. In addition to including a patient representative who provides lived experience with a particular disease, condition, or medical product, all advisory committee meetings include an open public hearing session during which patients and their caregivers have an opportunity to share relevant information about a given drug and disease or condition.39 In principle, open public hearing sessions should inform the advisory committee on the particular drug product under consideration. In practice, information shared may not be relevant or balanced.
Given limited time, speakers are typically allotted 5–10 minutes each to share their perspective with the advisory committee. Selection of the speakers is typically done on a lottery basis, which means that anyone may have the opportunity to share a point of view whether or not they have relevant experience with a given disease or the drug product being discussed (FDA, 2013b). For example, at the advisory committee meeting for Elevidys for Duchenne muscular dystrophy (DMD), of the 18 public commenters, 8 were not patients or their caregivers (FDA, 2023a). For rare disease patients and their families who have worked hard to reach this point and who depend on the expertise and evidence-based consideration of advisory committees, it can be defeating to share equal time with people who do not have relevant experience or expertise.
In keeping with patient focused drug development, FDA has an opportunity to shape open public hearings so that they provide better input from patients on clinically meaningful benefit and risk tolerance. This is particularly the case for drug development for rare diseases and conditions—patients are often the experts on the disease because, so little is known. Providing a more structured format for selecting speakers who have lived experience with a rare disease or condition or have experience with a given treatment and managing the session to ensure that speakers are contributing valuable information that is relevant to the marketing authorization application may enable a more focused and streamlined approach for seeking public input. Furthermore, better engagement and inclusion of people with lived experience throughout the regulatory lifecycle could help to ensure that the regulatory process is meeting the needs of patients with rare diseases.
A 2013 guidance for the public, FDA advisory committee members, and FDA staff specifies information that speaker requests should include name and affiliation, contact information, and a general description of the nature of the presentation (FDA, 2013b). It does not indicate that speakers should have firsthand experience with a given disease or condition or with the product under consideration. For rare disease patients and caregivers with lived experience, FDA should provide materials on the types of insights and perspectives that would be most beneficial for the advisory committee. This may include reference to relevant PFDD listening sessions or data relevant to the disease indication. For example, speakers could be asked to demonstrate how the primary or secondary outcomes relate to their ability to perform activities of daily living or other challenges or limitations they perceive of these measures.
Advisory committees should be well informed by the community of people most affected by the disease, and they need to listen to the patient voice when making evaluations about a drug to treat a rare disease or condition. In addition to speaking at an open hearing sessions, there are opportunities for patients and caregivers to submit written or electronic comments through the Federal Register. Taken together, these mechanisms are insufficient compared to the opportunity that FDA and the applicant have to speak with the advisory committee, each of which are guaranteed time on the agenda for formal presentation, including a question-and-answer period. Patients and their caregivers should have this dedicated allotted time as well, so their insights and perspectives can be weighted similarly to those of other key stakeholders at the table.
Conclusion 2-3: The strategic engagement of people who have rare diseases and their caregivers with lived experience throughout the drug review and approval process can help regulatory agencies facilitate the development of drug products that meet the needs of patients living with rare diseases and conditions.
RECOMMENDATION 2-2: The U.S. Food and Drug Administration (FDA) should strengthen mechanisms to integrate input from people living with a rare disease or condition, their caregivers, and patient representatives, especially patient groups that are small and under-resourced, throughout the full continuum of the drug development process. To that end, FDA should take the steps necessary to fully implement Section 1137 of the Food and Drug Administration Safety and Innovation Act (Public Law 112-144), which directs the Secretary of Health and Human Services to develop and implement strategies to solicit the views of rare disease patients during the full range of regulatory review discussions. This should include but not be limited to:
Sponsor Engagement
Sponsors developing new drugs must navigate a range of complex challenges when designing and conducting a study for regulatory submission. Clinical trials for regulatory submission require a combination of clinical, safety, biostatistical, and regulatory expertise, as well an understanding of the patient populations a drug is intended to treat to maximize the likelihood that study results meet regulatory requirements to gain market approval (FDA, 2017a). These challenges are heightened when it comes to rare diseases and conditions. Additionally, many companies developing rare disease drugs are small and medium-sized enterprises, which may have fewer resources and less in-house expertise than large pharmaceutical companies. For these reasons it is critically important for sponsors developing rare disease drug products to engage with the agency early and often.
Sponsors may request a meeting with FDA at any time during drug development; FDA may communicate with a sponsor at any time during the process about the need for more data or information. Regulatory project managers (RPMs) located within the clinical review divisions serve an important role as the main point of contact between FDA and sponsors, while nonclinical review division RPMs focus on specific issues (e.g., proprietary names, manufacturing, and controls) (FDA, 2017a).
Sponsors are encouraged to contact FDA through the appropriate RPM, and they are strongly discouraged from directly contacting reviewers assigned to their IND. The 2017 guidance for industry and review staff, Best Practices for Communication Between IND Sponsors and FDA During Drug Development, states, “CDER and CBER strive to provide timely and accurate advice and feedback to sponsors that represent the review teams’ current thinking on the issue, and this is best accomplished by adhering to the communication procedures described above and throughout this guidance” (FDA, 2017a). When appropriate, sponsors may solicit advice on issues including regulatory, clinical and statistical, safety, pharmacology and pharmacokinetics, toxicology, and product quality. In addition to communications between the sponsor and the RPM, sponsors may also request a formal meeting; these can be particularly useful at critical junctures or “milestone meetings” (FDA, 2017a).
Conclusion 2-4: FDA engagement with rare disease drug development sponsors is of particular importance because compared with common diseases, rare diseases are less well understood, more often do not have regulatory precedent, and more commonly lack validated endpoints and outcome measures and involve small patient populations which limit the size and number of clinical trials that can be conducted.
As discussed above, there are opportunities to better ensure that sponsors and other stakeholders, particularly small companies, have access to the knowledge, expertise, and tools to advance new drugs for the treatment of rare diseases and conditions.
RECOMMENDATION 2-3: The U.S. Food and Drug Administration and the National Institutes of Health in collaboration with the European Medicines Agency, nongovernmental organizations, patient groups, and biopharmaceutical sponsors, should implement a sponsor, investigator, and patient group navigation service to support the development of drugs to treat rare diseases and conditions (1) by advising on the range of available regulatory pathways and flexibilities and (2) by providing clarity on how to comply with regulatory policies, apply guidances, and meet requirements in rare disease drug development. Actions should include:
SELECT RARE DISEASE PROGRAMS
In addition to its programs focused on the approval process for rare disease products, FDA has several other programs relevant to rare diseases. These programs, several of which are in pilot form, are described below.
The Rare Disease Innovation Hub
In 2024, FDA announced a plan to establish a Rare Disease Innovation Hub (the Hub) to establish a new model for FDA to leverage cross-agency expertise and enhance shared learnings to spur drug development for rare diseases and conditions (FDA, 2024i). The Hub, which will be co-led by the director of CDER and the director of CBER, will provide services across all rare diseases with a special focus on treatments of smaller populations where natural history and disease progression is not fully understood (FDA, 2024i). This will include three primary functions:
- “Serve as a single point of connection and engagement with the rare disease community, including patient and caregiver groups, trade organizations, and scientific/academic organizations, for matters that intersect CDER and CBER. The Hub will help the larger rare disease community navigate important intersections across FDA that affect patients with rare diseases, such as medical devices, including diagnostic tests, and combination products.
- “Enhance intercenter collaboration to address common scientific, clinical and policy issues related to rare disease product development, including relevant cross-disciplinary approaches related to product review, and promote consistency across offices and Centers.
- “Advance regulatory science with dedicated workstreams for consideration of novel endpoints, biomarker development and assays, innovative trial design, real world evidence, and statistical methods” (FDA, 2024i).
In addition to enabling more collaboration across ongoing FDA rare disease programs, the Hub will seek to expand collaborative opportunities with patient groups, sponsors, researchers, and other interested parties (FDA, 2024i). Patients will have an opportunity to shape the Hub’s priorities through and open public meeting that will take place in the fall of 2024.
Accelerating Rare Disease Cures Program
CDER launched the Accelerating Rare disease Cures (ARC) Program in 2022; its mission is “to drive scientific and regulatory innovation and engagement to accelerate the availability of treatments for patients with rare diseases” (FDA, 2024c). The program is governed by senior leaders from the Office of New Drugs, the Office of the Center Director, and the Office of Translational Science and managed by CDER’s Rare Diseases Team; the Rare Diseases Team, established after the fifth reauthorization of PDUFA, is committed in the sixth and seventh reauthorizations of PDUFA to connect rare disease initiatives and programs across CDER. In addition to these groups that lead the ARC program, many other offices, divisions, and programs within FDA contribute and are connected to the work.
In the first year since its inception, ARC focused on stakeholder outreach and education, including FDA patient listening sessions, quarterly newsletters, and new initiatives aimed at gathering stakeholder input and making the rare disease drug approval process more transparent (FDA, 2023b). ARC launched its Learning and Education to Advance and Empower Rare Disease Drug Developers (LEADER 3D) initiative in 2023. Through LEADER 3D, FDA seeks input from stakeholders who design and conduct rare disease drug development programs in order to identify gaps in knowledge about the regulatory process. Input is curated and documented in a public report by FDA (FDA, 2024j) and will be used to create or expand educational resources for stakeholders. Also in 2023, ARC added two new filters to CDER’s Drugs and Biologics Dashboard hosted on FDA-TRACK (FDA, 2024a). This dashboard serves as a management program to report on performance measures and key projects at FDA. The new filters, “Original Rare Disease Application Approval” and “Novel Rare Disease Drugs Approval,” allow users to view information about the development and approval of products for rare diseases (FDA, 2023b).
In addition to these stakeholder-focused efforts, the ARC program has also supported scientific and regulatory initiatives, including the development of platforms to facilitate natural history studies, exploring methodologies to construct novel endpoints, the expansion of the use of drug/disease modeling; establishing efficient approaches to dose-selection for drugs for small population diseases, exploring innovative methodologies to trial design and interpretation for very small populations, and expanding efforts to support in translational medicine approaches for individual rare disease programs (FDA, 2023b).
Rare Disease Endpoint Advancement Pilot Program
The Rare Disease Endpoint Advancement (RDEA) Pilot Program is a joint CDER and CBER program that is intended to advance rare disease drug development by providing a mechanism for sponsors to collaborate with FDA throughout the efficacy endpoint development process. Sponsors submit a proposal for development of a novel efficacy endpoint for consideration to the RDEA pilot program; proposed endpoints are eligible for selection into the program if:
- The associated drug or biological product development program is active and addresses a rare disease (or a common disease where the endpoint or methodology could be applicable to a rare disease);
- The associated drug or biological product has an active IND or pre-IND for this treatment (with the exception of the endpoint being studied through natural history studies); and
- The proposed endpoint is a novel efficacy endpoint intended to establish substantial evidence of effectiveness for rare disease treatment. A novel endpoint is one that has never been used to support drug development or one that has been modified from its prior use (FDA, 2024p).
Announced in October 2022, the RDEA pilot will accept proposals through fiscal year 2027. When a proposal is admitted into the RDEA pilot program, FDA conducts an initial meeting and up to three followup meetings with the sponsor. At these meetings, sponsors can engage in focused discussion with interdisciplinary FDA experts and talk through the development of novel efficacy endpoints intended to establish evidence of effectiveness (FDA, 2023k). The RDEA meetings are in addition to other interactions with FDA that are conducted through the IND submission process.
The RDEA pilot program is intended to serve an educational purpose and encourage transparency and collaboration among the rare disease community (Lee et al., 2023). To this end, FDA conducts public workshops to discuss topics related to endpoint development for rare disease and can share information about the novel endpoints developed through the RDEA pilot program in various public-facing materials, such as in guidance documents and on the RDEA pilot program webpage. Sponsors who submit proposals to the RDEA pilot program for novel endpoints must agree to disclose certain elements of their endpoint development program prior to a product’s approval.
Support for clinical Trials Advancing Rare disease Therapeutics
Support for clinical Trials Advancing Rare disease Therapeutics (START) is a joint CBER and CDER pilot program that was launched in late 2023. It augments currently available formal meetings by addressing issues through more rapid, ad hoc communication mechanisms (FDA, 2023o; Lee et al., 2023). To be eligible, a drug development program must be under an active IND that is in electronic common technical document (eCTD) format, unless the IND is of a type granted a waiver from eCTD format and must have a chemistry manufacturing and controls development strategy that aligns with clinical development plans. For CBER-regulated products, the product must be a cell or gene therapy under IND being developed toward a BLA and must be intended to address an unmet medical need as a treatment for a rare disease or serious condition that is likely to lead to significant disability or death within the first decade of life. For CDER-regulated products, the product must be intended to treat rare neurodegenerative conditions, including those of rare genetic metabolic etiology (FDA, 2023o).
Sponsors who are selected to participate in START will receive enhanced communications with FDA review staff, including an initial meeting to review features of the pilot, discuss a pathway to support a marketing application, and to identify specific issues about which the sponsor would like further communication (FDA, 2023o). Additional communications may be conducted via email or teleconference, which will be scheduled or held as needed as agreed upon by the sponsor and FDA. The program is designed to be milestone-driven; that is, it is intended to help a product reach a significant regulatory milestone, such as initiation of a pivotal clinical study stage or the pre-BLA or pre-NDA meeting stage (FDA, 2023o). The additional communication provided to sponsors is intended to address issues that would otherwise delay or prevent a promising product from progressing along the pathway toward approval (FDA, 2023o).
Complex Innovative Trial Design Meeting Program
Due to the complex biology underpinning rare diseases and conditions, low disease prevalence, and patient heterogeneity, it is often challenging to design traditional randomized controlled trials for studying drugs that treat rare diseases or conditions. As such, clinical trials for rare diseases and conditions are often smaller than for other more prevalent conditions and may require the use of novel design elements to meet evidentiary standards. To assist sponsors, FDA’s Complex Innovative Trial Design Meeting program (also referred to as the Complex Innovative Trial Paired Meeting Program) offers sponsors up to two meetings with FDA to discuss their proposed complex innovative design elements during late-stage clinical development. In guidance, FDA explains that there is no fixed definition of a complex innovative design because what is considered innovative may change over time, and that the determination of whether a specific novel design is appropriate for regulatory use is made on a case-by-case basis (FDA, 2020a). The guidance provides examples of innovative design approaches (e.g., adaptive designs, Bayesian inference), and gives sponsors suggestions on common elements that should be included in a proposal for this program (FDA, 2020a). Like RDEA, the Complex Innovative Trial Design (CID) meeting program has educational components and requires sponsors to sign a disclosure agreement so that certain information can be shared publicly. FDA held several meetings on CIDs on diseases during which FDA and sponsors discussed trial design elements such as:
- Use of information from historical studies to increase the study power;
- Use of an active-controlled non-inferiority design;
- Model-based extrapolation from adults to the pediatric population; and
- Use of Bayesian methods and response adaptive randomization (FDA, 2023d).
In the first year of its existence, the CID meeting program selected five meeting requests, all of which were related to the use of Bayesian methods. Two of the requests were from sponsors developing products for rare diseases: Duchenne muscular dystrophy and pediatric multiple sclerosis (Price and Scott, 2021). Other CID meetings that supported rare disease drug product development focused on pediatric multiple sclerosis, lupus, epilepsy with myoclonic-atonic seizures (FDA, 2023d).
Bespoke Gene Therapy Consortium
FDA serves an advisory role in the Accelerating Medicines Partnership® Program Bespoke Gene Therapy Consortium, which is a public–private partnership aimed at facilitating the development of adeno-associated virus (AAV) gene therapies for individuals or small populations with very rare diseases.40 The consortium is involved in two concurrent and interrelated projects aimed at making AAV technology more broadly applicable and advancing scientific and regulatory approaches to AAV gene therapies:
- 1.
Further the understanding of AAV technology for gene therapy by supporting research in areas including enhancing vector generation and transgene expression for AAV gene therapies. Though AAV has been successfully used to treat genetic diseases, there is a need to better understand the underlying biology and how to best leverage this tool.
- 2.
Develop a standard operational playbook for developing these “bespoke” gene therapies for very rare diseases. The playbook will contain approaches to streamlining product development and navigating the regulatory pathway and standardized submission package templates to create a repeatable process for the development and regulatory approval of AAV gene therapies.
The first version of the playbook was released in February 2024 and focused on potential for streamlining pre-clinical and product testing, navigating the regulatory pathway, and standardized regulatory submissions (Bespoke Gene Therapy Consortium, 2024). The playbook serves as a guide for developers, with notes about best practices, decision trees to guide the submission process, and templates for submission requirements.
Opportunities for Expanding FDA Rare Disease Programs
Although the programs described above are welcome developments, most remain limited in scope and scale compared with the vastness of need in rare disease drug development. Some programs have existed for enough time to assess their effectiveness and strengths and weaknesses. Several programs are still early on in their implementation, making it difficult to assess their impact on orphan product development. For instance, the RDEA pilot as conceived will address only an exceedingly small fraction of the endpoint challenges that exist in rare disease development. Furthermore, while the START pilot program holds great promise, participant selection was only just publicly announced earlier this year, and, as such, the committee was unable to assess the program’s effectiveness or impact on orphan approvals.
Each of these special programs is intended to strengthen the overall orphan drug development and review, but their recency and proliferation present analytical challenges for program assessment. The lack of a track record makes it challenging for sponsors to choose the best path forward, increasing the complexity of designing an orphan drug development program.
Recommendation 2-4: The U.S. Food and Drug Administration (FDA) should assess the impact of new and ongoing programs and approaches that support drug development for rare diseases and conditions to improve the regulatory decision-making process; publicly share the results of these assessments in a timely manner; take steps to ensure that lessons learned across different programs are disseminated throughout FDA centers and divisions, including a summary of regulatory flexibilities and novel innovative approaches that were considered acceptable; scale up and expand successful programs across therapeutic areas; and modify or sunset programs that are not improving the regulatory decision-making process. Programs and regulatory approaches should include, but not be limited to:
TRANSPARENCY
In addition to public meetings, guidance documents, and other materials on regulatory policy, FDA makes certain information about approved drugs available on its public website Drugs@FDA.41 The database has records for drugs approved as early as 1939, but it does not contain FDA-approved products that are regulated by CBER. However, external parties can access records of approvals for CBER-regulated products via the Licensed Biological Products with Supporting Documents database. The availability of review documents can vary by drug. While review documents are helpful for third parties to understand FDA decision making, they are redacted to protect confidential information. There are several elements of medical product submissions and regulatory decisions that are not publicly released, including:
- Filing of an application;
- Patient-level datasets, clinical study reports, or other post-marketing reports associated with an application;
- Justification for orphan designation;
- Rationale for complete response;
- Safety or efficacy reasons for a clinical hold;
- Key milestones in product approval; and
- Underlying decisions for non-approval (Sharfstein et al., 2017).
While the elements above are not released, when the agency holds an advisory committee meeting, minutes of the meeting and commentary on the sponsors application are made public (Sharfstein et al., 2017).
SUMMARY OF CONCLUSIONS AND RECOMMENDATIONS
Conclusion 2-1: While the statutory requirements for drug approval for rare diseases and conditions are the same as for non-rare diseases or conditions, FDA has long recognized the need to apply regulatory flexibility in the review and approval of marketing authorization applications.
Conclusion 2-2: FDA is using available mechanisms to gather patient input. However, there are opportunities to better ensure that patient input informs the development of treatments for rare diseases as well as the design and conduct of clinical trials for rare diseases. More clarity is needed on the part of patient groups and people with lived experience on how the agency is using patient input to inform regulatory decision-making and what types of patient input are most relevant.
Conclusion 2-3: The strategic engagement of people who have rare diseases and their caregivers with lived experience throughout the drug review and approval process can help regulatory agencies facilitate the development of drug products that meet the needs of patients living with rare diseases and conditions.
Conclusion 2-4: FDA engagement with rare disease drug development sponsors is of particular importance because compared with common diseases, rare diseases are less well understood, more often do not have regulatory precedent, and more commonly lack validated endpoints and outcome measures and involve small patient populations which limit the size and number of clinical trials that can be conducted.
RECOMMENDATION 2-1: Congressional action is needed to encourage and incentivize more studies that provide information about the use of rare disease drug products in pediatric populations. To that end, Congress should remove the Pediatric Research Equity Act orphan exemption and require an assessment of additional incentives needed to spur the development of drugs to treat rare diseases or conditions.42
Additionally, the U.S. Food and Drug Administration (FDA) and the National Institutes of Health in partnership with nongovernmental organizations, including patient groups, clinical investigators, and biopharmaceutical companies, should work to provide clarity regarding the evolving regulatory policies and practices for the inclusion of pediatric populations as early as possible in rare disease clinical trials. Actions should include, but are not limited to the following:
RECOMMENDATION 2-2: The U.S. Food and Drug Administration (FDA) should strengthen mechanisms to integrate input from people living with a rare disease or condition, their caregivers, and patient representatives, especially patient groups that are small and under-resourced, throughout the full continuum of the drug development process. To that end, FDA should take the steps necessary to fully implement Section 1137 of the Food and Drug Administration Safety and Innovation Act (Public Law 112-144), which directs the Secretary of Health and Human Services to develop and implement strategies to solicit the views of rare disease patients during the full range of regulatory review discussions. This should include but not be limited to:
RECOMMENDATION 2-3: The U.S. Food and Drug Administration and the National Institutes of Health in collaboration with the European Medicines Agency, nongovernmental organizations, patient groups, and biopharmaceutical sponsors, should implement a sponsor, investigator, and patient group navigation service to support the development of drugs to treat rare diseases and conditions (1) by advising on the range of available regulatory pathways and flexibilities and (2) by providing clarity on how to comply with regulatory policies, apply guidances, and meet requirements in rare disease drug development. Actions should include:
RECOMMENDATION 2-4: The U.S. Food and Drug Administration (FDA) should assess the impact of new and ongoing programs and approaches that support drug development for rare diseases and conditions to improve the regulatory decision-making process; publicly share the results of these assessments in a timely manner; take steps to ensure that lessons learned across different programs are disseminated throughout FDA centers and divisions, including a summary of regulatory flexibilities and novel innovative approaches that were considered acceptable; scale up and expand successful programs across therapeutic areas; and modify or sunset programs that are not improving the regulatory decision-making process. Programs and regulatory approaches should include, but not be limited to:
REFERENCES
- Allen HC, Garbe MC, Lees J, Aziz N, Chaaban H, Miller JL, Johnson P, DeLeon S. Off-label medication use in children, more common than we think: A systematic review of the literature. Journal of the Oklahoma State Medical Association. 2018;111(8):776–783. [PMC free article: PMC6677268] [PubMed: 31379392]
- Anatol R. Regulatory flexibilities for cell and gene therapies for rare diseases: Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 3 - Virtual). 2024.
- Asbury CH. The Orphan Drug Act: The first 7 years. JAMA. 1991;265(7):893–897. [PubMed: 1992188]
- Benjamin DJ, Lythgoe MP. Modernising the US FDA’s accelerated approval pathway. Lancet Oncology. 2023;24(3):203–205. [PubMed: 36858720]
- Bespoke Gene Therapy Consortium. Regulatory playbook version 1.0. 2024. [August 1, 2024]. https://fnih
.org/wp-content /uploads/2024 /04/BGTC-Regulatory-Playbook-Version-1 .0.pdf . - Biotechnology Innovation Organization. FDA—sponsor engagement framework: Clinical outcomes assessments. 2022. [August 15, 2024]. https://www
.bio.org/sites /default/files/2023-06 /BIO_FDA_Sponsor _Engagement_Framework _for_COA_Development.pdf . - Brennan Z. Endpoints News. 2024. [February 27, 2024]. Accelerated approval will be ‘the norm’ for gene therapies, FDA’s Peter Marks says. https://endpts
.com/accelerated-approval-will-be-the-normfor-gene-therapies-fdas-peter-marks-says/ - Bugin Kn. U.S. Food and Drug Administration. d. . [August 1, 2024]. What does FDA do during review? How do we use the patient voice during review? https://www
.fda.gov/media /176607/download . - CIRS (Centre for Innovation in Regulatory Science). Data analysis commissioned by the Committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union. National Academies of Sciences, Engineering, and Medicine; Washington, DC: 2024. Data analysis and summary to help inform the National Academies committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union.
- Collins G, Stewart M, McKelvey B, Stires H, Allen J. Breakthrough therapy designation criteria identify drugs that improve clinical outcomes for patients: A case for more streamlined coverage of promising therapies. Clinical Cancer Research. 2023;29(13):2371–2374. [PMC free article: PMC10324637] [PubMed: 35616593]
- Committee on Drugs. Neville KA, Frattarelli DAC, Galinkin JL, Green TP, Johnson TD, Paul IM, Van Den Anker JN. Off-label use of drugs in children. Pediatrics. 2014;133(3):563–567. [PubMed: 24567009]
- Committee on Oversight and Accountability: U.S. House of Representatives. Testimony of Dr. Robert Califf. 2024. [August 1, 2024]. https://oversight
.house .gov/wp-content/uploads /2024/04/FDA-House-Oversight-and-Accountability-Testimony.pdf . - Critical Path Institute. Biomarker data repository. [August 15, 2024]. n.d. https://c-path
.org/program /biomarkerdata-repository/ - CTTI (Clinical Trials Transformation Initiative). Patient Engagement Collaborative. 2023. [March 10, 2024]. https:
//ctti-clinicaltrials .org/wp-content /uploads/2023/05/PEC-Framework _CompliantApr-10-2023_FINAL .pdf . - Donoghue M. Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 3 - Virtual). 2024. Rare Cancer Drug Development.
- Eastern Research Group Inc. Assessment of the use of patient experience data in regulatory decision-making. 2021. [August 1, 2024]. https://www
.fda.gov/media /150405/download?attachment . - Eglovitch J. FDA eyes collaborative review pilot for gene therapies. 2024. [January 12, 2024]. https://www
.raps.org /news-and-articles/news-articles /2024/1/fda-eyes-collaborative-review-pilot-for-genethera . - Epps C, Bax R, Croker A, Green D, Gropman A, Klein AV, Landry H, Pariser A, Rosenman M, Sakiyama M, Sato J, Sen K, Stone M, Takeuchi F, Davis JM. Global regulatory and public health initiatives to advance pediatric drug development for rare diseases. Therapeutic Innovation & Regulatory Science. 2022;56(6):964–975. [PMC free article: PMC9040360] [PubMed: 35471559]
- Fain K. FDA approval standards for new drugs and biological products: Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 1 - Virtual). 2023.
- FDA (U.S. Food and Drug Administration). Providing clinical evidence of effectiveness for human drug and biological product: Guidance for industry. 1998. [August 1, 2024]. https://www
.fda.gov/media/71655/download . - FDA. How to comply with the Pediatric Research Equity Act. 2005. [August 1, 2024]. https://www
.fda.gov/media/72274/download . - FDA. Applications for approval to market a new drug; complete response letter; amendments to unapproved applications. Federal Register. 2008;73(133) [August 1, 2024]; https://www
.govinfo.gov /content/pkg/FR-2008-07-10 /pdf/E8-15608.pdf . [PubMed: 18850675] - FDA. FDA report: Ensuring access to adequate information on medical products for all - with a special focus on underrepresented subpopulations, including racial subgroups. 2013a. [August 1, 2024]. https://www
.fda.gov/media/86023/download . - FDA. The open public hearing at FDA advisory committee meetings: Guidance for the public, FDA advisory committee members, and FDA staff. 2013b. [August 1, 2024]. https://www
.fda.gov/media/79874/download . - FDA. CDER MAPP 6030.2 Rev. 1: INDs: Review of Informed Consent Documents. 2014a. [August 1, 2024]. https://www
.fda.gov/media/72752/download . - FDA. Expedited programs for serious conditions—drugs and biologics: Guidance for industry. 2014b. [August 1, 2024]. https://www
.fda.gov/media/86377/download . - FDA. Advisory committees: Critical to the FDA’s product review process. 2016. [July 11, 2024]. https://www
.fda.gov/drugs /information-consumers-and-patients-drugs /advisory-committees-critical-fdas-product-review-process . [PubMed: 15032199] - FDA. Best practices for communication between IND sponsors and FDA during drug development guidance for industry and review staff: Good review practice. 2017a. [August 1, 2024]. https://www
.fda.gov/media/94850/download . - FDA. Orphan Drug Modernization Plan. 2017b. [March 19, 2024]. https://www
.fda.gov/industry /designatingorphan-product-drugs-and-biological-products /orphan-drug-modernization-plan . - FDA. Clarification of orphan designation of drugs and biologics for pediatric subpopulations of common diseases: Guidance for industry. 2018a. [March 19, 2024]. https://www
.fda.gov/media /109496/download . - FDA. Fact sheet: Pediatric provisions in the Food and Drug Administration Safety and Innovation Act (FDASIA). 2018b. [August 1, 2024]. https://www
.fda.gov/regulatory-information /food-and-drugadministration-safety-and-innovation-act-fdasia /fact-sheet-pediatric-provisions-food-and-drug-administration-safety-and-innovation-act-fdasia . - FDA. Information sharing. 2018c. [August 15, 2024]. https://www
.fda.gov/federal-state-local-tribal-and-territorial-officials /communications-outreach /information-sharing . - FDA. Demonstrating substantial evidence of effectiveness for human drug and biological products: Guidance for industry. 2019a. [August 1, 2024]. https://www
.fda.gov/media /133660/download . - FDA. Expedited programs for regenerative medicine therapies for serious conditions: Guidance for industry. 2019b. [August 1, 2024]. https://www
.fda.gov/media /120267/download . - FDA. FDA takes first action under new international collaboration with Australia and Canada designed to provide a framework for concurrent review of cancer therapies, approving treatment for patients with endometrial carcinoma: News release. 2019c. [August 1, 2024]. https://www
.fda.gov/news-events /press-announcements /fda-takes-first-action-under-new-international-collaboration-australia-and-canada-designed-provide . - FDA. Rare pediatric disease priority review vouchers guidance for industry. 2019d. [August 1, 2024]. https://www
.fda.gov/media/90014/download . - FDA. Interacting with the FDA on complex innovative trial designs for drugs and biological products: Guidance for industry. 2020a. [August 1, 2024]. https://www
.fda.gov/media /130897/download . - FDA. Pediatric study plans: Content of and process for submitting initial pediatric study plans and amended initial pediatric study plans: Guidance for industry. 2020b. [August 1, 2024]. https://www
.fda.gov/media/86340/download . - FDA. Action package for posting SOPP 8401.7. 2022a. [August 1, 2024]. https://www
.fda.gov/media/82426/download . - FDA. Development & approval process | drugs. 2022b. [June 12, 2024]. https://www
.fda.gov/drugs /development-approval-process-drugs . - FDA. Enhancing benefit-risk assessment in regulatory decision-making. 2022c. [August 15, 2024]. https://www
.fda.gov/industry /prescription-drug-user-fee-amendments /enhancing-benefit-risk-assessment-regulatory-decision-making . - FDA. Ethical considerations for clinical investigations of medical products involving children. 2022d. [August 1, 2024]. https://www
.fda.gov/media /161740/download . - FDA. Externally-led patient-focused drug development meetings. 2022e. [August 7, 2024]. https://www
.fda.gov/industry /prescription-drug-user-fee-amendments /externally-led-patient-focused-drugdevelopment-meetings . - FDA. General clinical pharmacology considerations for neonatal studies for drugs and biological products: Guidance for industry. 2022f. [August 1, 2024]. https://www
.fda.gov/media /129532/download . - FDA. General clinical pharmacology considerations for pediatric studies of drugs, including biological products. 2022g. [August 1, 2024]. https://www
.fda.gov/media/90358/download . - FDA. Project Orbis frequently asked questions. 2022h. [August 1, 2024]. https://www
.fda.gov/about-fda /oncology-center-excellence /project-orbis-frequently-asked-questions . - FDA. 74th Cellular, Tissue, and Gene Therapies Advisory Committee (CTGTAC). 2023a. [August 1, 2024]. https://www
.fda.gov/media /169105/download . - FDA. Accelerating Rare disease Cures (ARC) Program: Year one anniversay update. 2023b. [August 1, 2024]. https://www
.fda.gov/media /169429/download . - FDA. Benefit-risk assessment for new drug and biological products: Guidance for industry. 2023c. [August 1, 2024]. https://www
.fda.gov/media /152544/download . - FDA. Complex Innovative Trial Design meeting program. 2023d. [March 19, 2024]. https://www
.fda.gov/drugs /development-resources /complex-innovative-trial-design-meeting-program . - FDA. Considerations for long-term clinical neurodevelopmental safety studies in neonatal product development. 2023e. [August 1, 2024]. https://www
.fda.gov/media /165239/download . - FDA. Demonstrating substantial evidence of effectiveness with one adequate and well-controlled clinical investigation and confirmatory evidence: Guidance for industry. 2023f. [August 1, 2024]. https://www
.fda.gov/media /172166/download . - FDA. FDA Rare Disease Day 2023: “Intersections with rare diseases—a patient focused event”. 2023g. [August 1, 2024]. https://www
.youtube.com /watch?v=ylk7eYTgUMM . - FDA. Frequently asked questions (FAQ) about designating an orphan product. 2023h. [March 19, 2024]. https://www
.fda.gov/industry /designating-orphan-product-drugs-and-biological-products /frequently-asked-questions-faq-about-designating-orphan-product . - FDA. Pediatric drug development under the Pediatric Research Equity Act and the Best Pharmaceuticals for Children Act: Scientific considerations. 2023i. [August 1, 2024]. https://www
.fda.gov/media /168202/download . - FDA. Pediatric drug development: Regulatory considerations—Complying with the Pediatric Research Equity Act and qualifying for pediatric exclusivity under the Best Pharmaceuticals for Children Act. 2023j. [August 1, 2024]. https://www
.fda.gov/media /168201/download . - FDA. Rare Disease Endpoint Advancement pilot program frequently asked questions. 2023k. [August 15, 2024]. https://www
.fda.gov/drugs /development-resources /rare-disease-endpoint-advancement-pilot-program-frequently-asked-questions . - FDA. Rare diseases: Considerations for the development of drugs and biological products: Guidance for industry. 2023l. [August 1, 2024]. https://www
.fda.gov/media /119757/download . - FDA. Real-time oncology review (RTOR): Guidance for industry. 2023m. [August 1, 2024]. https://www
.fda.gov/media /173641/download . - FDA. Regenerative medicine advanced therapy designation. 2023n. [July 19, 2024]. https://www
.fda.gov/vaccines-blood-biologics /cellular-gene-therapy-products /regenerative-medicine-advancedtherapy-designation . - FDA. Support for clinical Trials Advancing Rare disease Therapeutics pilot program; program announcement. 2023o. [August 1, 2024]. https://www
.federalregister .gov/documents /2023/10/02/2023-21235 /support-for-clinical-trials-advancing-rare-disease-therapeutics-pilot-program-program-announcement . - FDA. What we do. 2023p. [July 19, 2024]. https://www
.fda.gov/about-fda/what-we-do . - FDA. About FDA-TRACK. 2024a. [August 15, 2024]. https://www
.fda.gov/about-fda /fda-track-agency-wideprogram-performance /about-fda-track . - FDA. About the FDA patient representative program. 2024b. [March 10, 2024]. https://www
.fda.gov/patients /learn-about-fda-patient-engagement /about-fda-patient-representative-program . - FDA. Accelerating Rare disease Cures (ARC) program. 2024c. [February 20, 2024]. https://www
.fda.gov/aboutfda /center-drug-evaluation-and-research-cder /accelerating-rare-disease-cures-arc-program . - FDA. CDER patient-focused drug development. 2024d. [March 05, 2024]. https://www
.fda.gov/drugs /development-approval-process-drugs /cder-patient-focused-drug-development . - FDA. Condition-specific meeting reports and other information related to patients’ experience. 2024e. [August 15, 2024]. https://www
.fda.gov/industry /prescription-drug-user-fee-amendments /condition-specific-meeting-reports-and-other-information-related-patients-experience . - FDA. FDA-led Patient-Focused Drug Development (PFDD) public meetings. 2024f. [August 7, 2024]. https://www
.fda.gov/industry /prescription-drug-user-fee-amendments /fda-led-patient-focused-drug-development-pfdd-public-meetings . - FDA. FDA briefing document: Pediatric oncology subcommittee of the Oncologic Drugs Advisory Committee (ODAC). 2024g. [August 1, 2024]. https://www
.fda.gov/media /178681/download . - FDA. FDA patient listening sessions. 2024h. [August 7, 2024]. https://www
.fda.gov/patients /learn-about-fdapatient-engagement /fda-patient-listening-sessions . - FDA. FDA Rare Disease Innovation Hub to enhance and advance outcomes for patients. 2024i. [August 15, 2024]. https://www
.fda.gov/news-events /fda-voices /fda-rare-disease-innovation-hub-enhance-and-advance-outcomes-patients . - FDA. LEADER 3D: Learning and Education to Advance and Empower Rare Disease Drug Developers: Public report of external stakeholder analysis. 2024j. [August 15, 2024]. https://www
.fda.gov/media /176557/download?attachment= - FDA. Network of experts program: Connecting the FDA with external expertise. 2024k. [August 15, 2024]. https://www
.fda.gov/about-fda /center-devices-and-radiological-health /network-experts-program-connecting-fda-external-expertise . - FDA. Oncology Center of Excellence. 2024l. [May 29, 2024]. https://www
.fda.gov/about-fda /fda-organization /oncology-center-excellence . - FDA. Pediatric labeling changes. 2024m. [August 8, 2024]. https://www
.fda.gov/science-research /pediatrics /pediatric-labeling-changes . - FDA. Pediatric Research Equity Act. 2024n. [August 1, 2024]. https://www
.fda.gov/drugs /development-resources /pediatric-research-equity-act-prea . - FDA. Prescription Drug User Fee Amendments. 2024o. [June 25, 2024]. https://www
.fda.gov/industry /fda-userfee-programs /prescription-drug-user-fee-amendments . - FDA. Rare Disease Endpoint Advancement pilot program. 2024p. [August 6, 2024]. https://www
.fda.gov/drugs /development-resources /rare-disease-endpoint-advancement-pilot-program . - FDA. Rare pediatric disease designation and priority review voucher programs. 2024q. [August 15, 2024]. https://www
.fda.gov/industry /medical-products-rare-diseases-and-conditions /rare-pediatric-dis-ease-designation-and-priority-review-voucher-programs . - FDA. Best Pharmaceuticals for Children Act and Pediatric Research Equity Act - Status Report to Congress: July 1, 2015—June 30, 2020. [August 1, 2024]. n.d.-a. https://www
.fda.gov/media /157840/download . - FDA. Collecting patient experience data: How you can best help FDA. [August 1, 2024]. n.d.-b. https://www
.fda.gov/media /112163/download . - Fermaglich LJ, Miller KL. A comprehensive study of the rare diseases and conditions targeted by orphan drug designations and approvals over the forty years of the Orphan Drug Act. Orphanet Journal of Rare Diseases. 2023;18(1):163. [PMC free article: PMC10290406] [PubMed: 37353796]
- Fox TA, Booth C. Improving access to gene therapy for rare diseases. Disease Models & Mechanisms. 2024;17(6) [PMC free article: PMC11051979] [PubMed: 38639083]
- Freilich ER. Rare disease drug development: Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 3 - Virtual). 2024.
- Fung A, Yue X, Wigle PR, Guo JJ. Off-label medication use in rare pediatric diseases in the United States. Intractable & Rare Diseases Research. 2021;10(4):238–245. [PMC free article: PMC8630459] [PubMed: 34877235]
- GAO (U.S. Government Accountabilty Office). FDA’s priority review voucher programs. 2020. [August 1, 2024]. https://www
.gao.gov/assets/gao-20-251 .pdf . - GAO. Pediatric cancer studies: Early results of the Research to Accelerate Cures and Equity for Children Act. 2023. [August 1, 2024]. https://www
.gao.gov/products /gao-23-105947 . - Gao YG, Roberts S, Guy A. Maximizing regulatory review efficiency: The evolution of the FDA OCE RTOR pilot. Therapeutic Innovation & Regulatory Science. 2022 Oct;9(56)(2):212–219. [PMC free article: PMC8744371] [PubMed: 35006588]
- Hunter NL, O’Callaghan KM, Califf RM. Engaging patients across the spectrum of medical product development: View from the US Food and Drug Administration. JAMA. 2015;314(23):2499–2500. [PubMed: 26584067]
- Hwang TJ, Bourgeois FT, Franklin JM, Kesselheim AS. Impact of the priority review voucher program on drug development for rare pediatric diseases. Health Affairs. 2019;38(2):313–319. [PubMed: 30715972]
- ICH (International Council for Harmonsation of Technical Requirements for Pharmaceuticals for Human Use). Pediatric extrapolation - E11A. 2022. [August 1, 2024]. https://www
.fda.gov/media /161190/download . - IOM (Institute of Medicine). Addressing the barriers to pediatric drug development: Workshop summary. Vanchieri C, Butler AS, Knutsen A, editors. Washington, DC: The National Academies Press; 2008. [PubMed: 20669414]
- Kesselheim AS, Darrow JJ. FDA designations for therapeutics and their impact on drug development and regulatory review outcomes. Clinical Pharmacology & Therapeutics. 2015;97(1):29–36. [PubMed: 25670381]
- Kieffer CM, Miller AR, Chacko B, Robertson AS. Therapeutic Innovation & Regulatory Science. 2019. FDA reported use of patient experience data in 2018 drug approvals; p. 2168479019871519. [PubMed: 31597462]
- Kim T. Oncology Center for Excellence (OCE) regulatory programs. 2022. [August 1, 2024]. https://www
.fda.gov/media /165674/download . - Kimmel L, Conti RM, Volerman A, Chua KP. Pediatric orphan drug indications: 2010-2018. Pediatrics. 2020;145(4) [PMC free article: PMC7111489] [PubMed: 32127360]
- Kuehn CM. Patient experience data in US Food and Drug Administration (FDA) regulatory decision making: A policy process perspective. Therapeutic Innovation & Regulatory Science. 2018;52(5):661–668. [PubMed: 29714560]
- Lee KJ, Bent R, Vaillancourt J. Selected FDA programs on rare disease: Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 1 - Virtual). 2023.
- Lu C-F, Abbott CK. FDA takes first step toward international regulation of gene therapies to treat rare diseases. 2024. [July 19, 2024]. https:
//natlawreview .com/article/fda-takes-first-step-toward-international-regulation-gene-therapies-treat-rare#google _vignette . - Mease C, Miller KL, Fermaglich LJ, Best J, Liu G, Torjusen E. Analysis of the first ten years of FDA’s rare pediatric disease priority review voucher program: Designations, diseases, and drug development. Orphanet Journal of Rare Diseases. 2024;19(1):86. [PMC free article: PMC10895788] [PubMed: 38403586]
- Michaeli T, Jürges H, Michaeli DT. FDA approval, clinical trial evidence, efficacy, epidemiology, and price for non-orphan and ultra-rare, rare, and common orphan cancer drug indications: Cross sectional analysis. BMJ. 2023;381:e073242. [PMC free article: PMC10167557] [PubMed: 37160306]
- Miller KL, Lanthier M. Trends in orphan new molecular entities, 1983-2014: Half were first in class, and rare cancers were the most frequent target. Health Affairs (Millwood). 2016;35(3):464–470. [PubMed: 26953301]
- Monge AN, Sigelman DW, Temple RJ, Chahal HS. Use of US Food and Drug Administration expedited drug development and review programs by orphan and nonorphan novel drugs approved from 2008 to 2021. JAMA Network Open. 2022;5(11):e2239336. [PMC free article: PMC9627417] [PubMed: 36318210]
- Murray LT, Howell TA, Matza LS, Eremenco S, Adams HR, Trundell D, Coons SJ., Rare Disease Subcommittee of the Patient-Reported Outcome Consortium. Approaches to the assessment of clinical benefit of treatments for conditions that have heterogeneous symptoms and impacts: Potential applications in rare disease. Value Health. 2023;26(4):547–553. [PubMed: 36455827]
- NASEM (National Academies of Sciences, Engineering and Medicine). Living with ALS. Leshner AI, English RA, Alper J, editors. Washington, DC: The National Academies Press; 2024. [PubMed: 38917255]
- NICHD (Eunice Kennedy Shriver National Institute of Child Health and Human Development). About BPCA. [August 1, 2024]. n.d. https://www
.nichd.nih .gov/research/supported /bpca/about#:~:text=The %20latest%20renewal %20of%20the,6%20months %20of%20patent%20exclusivity . - NORD (National Organization for Rare Disorders). Impact of the rare pediatric disease priority review voucher program on drug development 2012 - 2024. 2024. [August 1, 2024]. https:
//rarediseases .org/wp-content/uploads /2024/07/NORD-Pediatric-PRV-Report .pdf . - OIG (U.S. Office of Inspector General). Delays in confirmatory trials for drug applications granted FDA’s accelerated approval raise concerns. 2022. [August 1, 2024]. https://oig
.hhs.gov/oei /reports/OEI-01-21-00401.pdf . - PhRMA (Pharmaceutical Research and Manufacturers of America). PREA and BPCA: Spurring pediatric drug development. 2020. [August 6, 2024]. https://phrma
.org/-/media /Project/PhRMA/PhRMAOrg /PhRMA-Org/PDF /P-R/PREA-and-BPCA-Doc-2022_2.pdf . - Poddar A, Raggio M, Concato J. Decisions on non-oncology breakthrough therapy designation requests in 2017-2019. Therapeutic Innovation & Regulatory Science. 2024;58(1):214–221. [PMC free article: PMC10764372] [PubMed: 37926768]
- Price D, Scott J. The U.S. Food and Drug Administration’s complex innovative trial design pilot meeting program: Progress to date. Clinical Trials. 2021;18(6):706–710. [PubMed: 34657476]
- Raggio M. FDA expedited programs: Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 1 - Virtual). 2023.
- Redberg RF. Faster drug approvals are not always better and can be worse. JAMA Internal Medicine. 2015;175(8):1398. [PubMed: 26098077]
- Sachs AN, Avant D, Lee CS, Rodriguez W, Murphy MD. Pediatric information in drug product labeling. JAMA. 2012;307(18):1914–1915. [PubMed: 22570457]
- Sasinowski FJ. Quantum of effectiveness evidence in FDA’s approval of orphan drugs. 2011. [August 1, 2024]. https://cdn
.rarediseases .org/wordpresscontent /wp-content/uploads /2014/12/NORDstudy-of-FDA-approval-of-orphan-drugs.pdf . [PubMed: 30227043] - Sasinowski FJ, Panico EB, Valentine JE. Quantum of effectiveness evidence in FDA’s approval of orphan drugs: Update, July 2010 to June 2014. Therapeutic Innovation & Regulatory Science. 2015;49(5):680–697. [PubMed: 30227043]
- Sharfstein JM, Miller JD, Davis AL, Ross JS, McCarthy ME, Smith B, Chaudhry A, Alexander GC, Kesselheim AS. Blueprint for transparency at the US Food and Drug Administration: Recommendations to advance the development of safe and effective medical products. The Journal of Law, Medicine & Ethics. 2017;45(2_suppl):7–23.
- Shea M, Ostermann L, Hohman R, Roberts S, Kozak M, Dull R, Allen J, Sigal E. Impact of breakthrough therapy designation on cancer drug development. Nature Reviews Drug Discovery. 2016;15(3):152. [PubMed: 26931085]
- Temkin E, Trinh J. FDA’s accelerated approval pathway: A rare disease perspective—history and opportunities for reform. 2021. [August 1, 2024]. https:
//rarediseases .org/wp-content/uploads /2022/10/NRD-2182-Policy-Report _Accelerated-Approval_FNL .pdf . - The Alliance for a Stronger FDA. Alliance webinar series: Webinar transcript with Nicole Verdun, head of the new CBER Office of Therapeutic Products. 2024. [August 1, 2024]. https://acrobat
.adobe .com/id/urn:aaid:sc :va6c2:8a91f106-2820-47a4-b90b-eef42686d6ff?viewer %21megaVerb=group-discover . - Treatment Action Group and Elizabeth Glaser Pediatric AIDS (acquired immunodeficiency syndrome) Foundation. Ensuring treatment for children with orphan diseases: Ending exemptions from the Pediatric Research Equity Act (PREA). 2019. [August 1, 2024]. https://www
.treatmentactiongroup .org/wp-content /uploads/2019 /08/prea_brief_2019_final.pdf . - Valentine JE, Sasinowski FJ. Orphan drugs and rare diseases. In: Piantadosi S, Meinert CL, editors. Principles and Practice of Clinical Trials. Cham: Springer International Publishing; 2020. pp. 1–19.
- Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: Diagnosing rare disease in children. Nature Reviews Genetics. 2018;19(5) [PubMed: 29398702]
Footnotes
- 1
Robert Califf, testimony to Committee on Oversight and Accountability. Available at https://oversight
.house .gov/wp-content/uploads /2024/04/FDA-House-Oversight-and-Accountability-Testimony.pdf (accessed June 26, 2024). - 2
P.L. 75–717. Federal Food, Drug, and Cosmetic Act (June 25, 1938).
- 3
P.L. 78–410. Public Health Service Act (July 1, 1944).
- 4
21 U.S.C. § 355(d).
- 5
21 P.L. 105–115. Food and Drug Modernization Act (November 21, 1997).
- 6
FDA develops guidance documents to share FDA’s current thinking on a topic (FDA, 2023f). These documents are not legally binding for FDA or the public. To develop a guidance document, FDA first has the option to seek input from external stakeholders. FDA then prepares a draft version of the document which is posted publicly along with a notice in the Federal Register. The draft guidance is then open to public comment. FDA then reviews the comments and makes any necessary edits before posting the finalized guidance document (21 CFR §10.115).
- 7
This sentence was edited after release of the prepublication version of the report to reflect the nature of data used.
- 8
This section was edited after release of the prepublication version of the report to clarify when a single trial approach could be sufficient for demonstrating effectiveness.
- 9
P.L. 112–144. Food and Drug Administration Safety and Innovation Act (July 9, 2012).
- 10
21 CFR §316.20(b)(8); 21 U.S.C. § 360bb(a)(2).
- 11
78 FR 35117(III)(A).
- 12
21 CFR §316.20(a).
- 13
78 FR 35117(III)(A) at 35119, explaining that: “orphan subset is a regulatory concept specific to the Orphan Drug regulations, and that it does not simply mean any medically recognizable or clinically distinguishable subset of persons with a particular disease or condition (as the term ‘medically plausible’ in this context may have been erroneously interpreted to imply).”
- 14
21 CFR §316.20(b)(6).
- 15
21 CFR §316.3(b)(13).
- 16
78 FR 35117(III)(A).
- 17
78 FR 35117(III)(A).
- 18
21 CFR §316.20.
- 19
This sentence was edited after release of the prepublication version of the report to more accurately reflect 21 USC § 360cc – Protection for drugs for rare diseases or conditions.
- 20
21 U.S.C §360cc(a).
- 21
21 U.S.C §360ee(b).
- 22
This section was edited after release of the prepublication version of the report to more accurately describe the data requested and received.
- 23
21 U.S.C. § 360ff.
- 24
21 U.S.C. § 360ff(a)(3).
- 25
21 U.S.C. § 360ff(d)(3).
- 26
21 U.S.C. § 360ff(b).
- 27
This sentence was edited after release of the prepublication version of the report to more accurately describe the accelerated approval process.
- 28
More information on the Critical Path Institute’s Biomarker Data Repository can be found at https://c-path
.org/program /biomarker-data-repository (accessed June 26, 2024). - 29
21 U.S.C. § 356b(1).
- 30
21 U.S.C. § 355f(g).
- 31
Human cells, tissues, and cellular and tissue-based products that are regulated solely under Section 361 of the Public Health Service Act and 21 CFR, part 1271 (aka “361 HCT/Ps”) do not undergo pre-market review. These products must meet the four criteria described in 21 CFR part 1271.10(a) (see https://www
.ecfr.gov /current/title-21/chapter-I /subchapter-L /part-1271/subpart-A/section-1271.10) (accessed March 16, 2024). - 32
The sentence has been modified after release of the prepublication version of the report to provide temporal context.
- 33
H.R.1231—RACE for Children Act—115th Congress (2017–2018).
- 34
This sentence was edited after release of the prepublication version of the report to clarify the intent of the recommendation.
- 35
21 U.S.C. 360bbb–8c.
- 36
This section was edited after release of the prepublication version of the report to more accurately reflect the article cited.
- 37
For more information, see https://www
.ema.europa .eu/en/events/meeting-ctti-fda-patient-engagement-collaborative-pec-and-ema-patients-and-consumers-working-party-pcwp-0 (accessed March 19, 2024). - 38
Title III, Section 3001(c) of the 21st Century Cures Act defines patient experience data as data that “(1) are collected by any persons (including patients, family members and caregivers of patients, patient advocacy organizations, disease research foundations, researchers, and drug manufacturers); and (2) are intended to provide information about patients’ experiences with a disease or condition, including—(A) the impact of such disease or condition, or a related therapy, on patients’ lives; and (B) patient preferences with respect to treatment of such disease or condition.’’
- 39
21 CFR §14.25(a).
- 40
This sentence was edited after release of the prepublication version of the report to more clearly describe FDA’s role.
- 41
Users can search by drug name or by month of approval, and can access information on drug name, active ingredients, patient labeling, marketing status, regulatory history, priority review and orphan designation status, and therapeutic equivalents. Available at https://www
.accessdata .fda.gov/scripts/cder/daf/index.cfm (accessed March 19, 2024). - 42
This sentence was edited after release of the prepublication version of the report to clarify the intent of the recommendation.
- DRUG REVIEW AND APPROVAL
- DESIGNATION FOR RARE DISEASE PRODUCTS
- EXPEDITED REGULATORY PROGRAMS
- Priority Review
- Regenerative Medicine Advanced Therapy
- Oncology Center of Excellence Programs
- Orphan Drugs and Expedited Review
- INCLUSION OF PEDIATRIC POPULATIONS
- STAKEHOLDER ENGAGEMENT
- SELECT RARE DISEASE PROGRAMS
- TRANSPARENCY
- SUMMARY OF CONCLUSIONS AND RECOMMENDATIONS
- REFERENCES
- FDA Flexibilities, Authorities, and Mechanisms - Regulatory Processes for Rare D...FDA Flexibilities, Authorities, and Mechanisms - Regulatory Processes for Rare Disease Drugs in the United States and European Union
Your browsing activity is empty.
Activity recording is turned off.
See more...