

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union; Shore CK, Worku TL, Smith CW, et al., editors. Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities. Washington (DC): National Academies Press (US); 2024 Oct 30.

Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities.
Show detailsINTRODUCTION
The purpose of the data analysis was to help inform the National Academies Committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union.1
The National Academies approached the Centre for Innovation in Regulatory Science (CIRS) to produce a commissioned data analysis and summary of key findings based on the marketing submissions, regulatory orphan designations, and marketing approvals of new active substances (NASs) to treat rare diseases and conditions by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA). The National Academies and CIRS entered into a contract agreement and agreed on the relevant data to be collected, definitions, and the analysis to be carried out by CIRS. CIRS undertook the data collection and prepared the analysis. The analysis was presented to the committee at regular meetings. The aim of those meetings was to provide feedback on the analysis, discuss the findings, and agree on additional analysis as well as next steps. Figure D-1 shows a graphical representation of the timeline of the project.
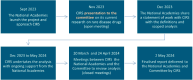
FIGURE D-1
Project timelines and steps. SOURCE: CIRS Data Analysis, 2024.
OVERALL SCOPE
The overall analysis was limited to initial marketing authorizations by EMA and FDA and focused on new active substances (NASs).2
Applications that were excluded from the data analysis:
- Vaccines
- Biosimilars
- Any other application where new clinical data were submitted
- Generic applications
- Applications for which a completely new dossier was submitted from a new company for the same indications as those already approved for another company
- Applications for a new or additional name, or a change of name, for an existing compound (i.e., a “cloned” application)
- Emergency use or special authorizations derived from an emergency (e.g., COVID-19 pandemic)
The analysis was divided into two main parts as outlined in Figure D-2: part A, analysis of approval rates (applications submitted versus approved), and part B, analysis of approved products.

FIGURE D-2
Overall methods and data sources used based on the contract agreement with the National Academies. SOURCE: CIRS Data Analysis, 2024.
As a result of the lack of available data in the public domain on part A (submissions) for FDA, this information was requested and obtained directly from the agency. Similarly, information on submissions to EMA was also obtained from the agency. Consequently, the data sources and products included differ when comparing parts A and B and are therefore described separately below.
For part B, data was retrieved from CIRS proprietary databases, which contains information extracted by CIRS from the public domain. Additional data points were also collected (Figure D-2).
PART A: ANALYSIS OF APPROVAL RATES
Product Scope
Products included in the analysis were NASs submitted by EMA (centralized procedure) or FDA (Center for Drug Evaluation and Research [CDER] and Center for Biologics Evaluation and Research [CBER]).
- Data obtained from FDA: New drug applications (NDAs) and biologics license applications (BLAs): Type 1 (drug product contains a new molecular entity) and Type 1,4 (combination drug product when at least one of the active ingredients is a new molecular entity).
- Data collected for EMA: Marketing authorization applications for a drug product that contains an NAS.
Caveat: These definitions differ slightly compared with the CIRS NAS definition used for Part B outlined below, resulting in a different set of products approved.
Year Ranges
Due to the fact that data provided by FDA were limited to 2015–2020, the same scope was applied to EMA in terms of the data collected from the agency.
Data Sources and Collection
FDA and EMA were approached by National Academies staff on behalf of the committee to request data relating to marketing authorization submissions and their corresponding regulatory outcomes. The following information was requested:
- Year range for analysis: 2013–2022
- Data categorization: All data broken down by year of submission cohort, orphan versus non-orphan designation, and therapeutic area following the World Health Organization Anatomical Therapeutic Chemical (ATC) classification system.
- Data requested
- Number of submitted NAS marketing authorization applications
- Number of applications that received marketing authorization
- Number of applications that were not under review
- Number of applications that were refused to file in the year of submission
- Number of applications that were withdrawn by the sponsor in the year of submission
- Number of applications that received a complete response letter or negative review
In response to these requests, FDA provided tabular outputs generated using data from DASH and RMS BLA databases and provided counts that met the following criteria:
- New molecular entities (NME), NDAs, and original BLAs received by CDER from January 1, 2015, to December 31, 2020.
- Original BLAs received by CBER from January 1, 2015, to December 31, 2020.
- Approval and non-approval actions (i.e., refuse to file, complete response, and withdrawals) as of December 31, 2023, stratified by the current (post-reorganization) Office of New Drugs (OND) review office and orphan designation.
- Approval and non-approval actions (i.e., refuse to file, complete response, and withdrawals as of December 31, 2023, stratified by three CBER-regulated biological product categories (i.e., cell and gene therapies; plasma-derived products; and other biological products) and orphan designation status.
- Median time to approval from FDA receipt date, regardless of filing status, is presented to account for multiple review cycles. There may be instances in which a submission received between January 1, 2015, and December 31, 2020, was reviewed more than once and did not receive an approval as of December 31, 2023. Those submissions are not included in the analysis of median time to approval from FDA receipt date.
FDA internal datasets were shared in confidence with CIRS (nonpublic data) under a contract agreement between the National Academies and CIRS. EMA provided some internal data extracts and a pivot table with links to publicly available information related to medicine that has been reviewed by the agency. Information was extracted and consolidated by CIRS.
Data Characteristics
For FDA, the following data were obtained from the agency:
- Number of applications approved by CDER and CBER between 2015 and 2020, broken down by year of submission and orphan status. CDER data was also broken down by FDA office, while CBER’s data was broken down by type of product.
- Number of applications not approved (including complete response [CR] letters,3 refusals to file and withdrawn applications) by CDER and CBER between 2015 and 2020 (lumped), broken down per orphan status. CDER data were also broken down by FDA office, while CBER’s data were broken down by type of product.
For EMA, the following data were extracted by CIRS from the agency’s websites:
- Databases with information on applications evaluated by EMA broken down as follows: product information (brand and generic name), orphan status, ATC code, milestone dates (validation, EMA Committee for Medicinal Products for Human Use opinion, EC decision date, etc.), type of outcome (approved, refused and withdrawn)
PART B: ANALYSIS OF APPROVED PRODUCTS
Recognizing the importance of advancing regulatory practices, CIRS has been benchmarking major regulatory agencies since 2002 using a methodology developed with the authorities (Hirako et al., 2007). The study continues today and focuses on new active substances approved by six regulatory agencies including FDA and EMA (CIRS, 2023). CIRS used its proprietary database, updated annually for the above-described study, in order to undertake the analysis of approved products. This database was supplemented with additional data points collected by CIRS as shown in Figure D-2.
Product Scope
Products included in the analysis were NASs approved by EMA (centralized procedure) or FDA (CDER and CBER).
NASs were defined by CIRS as a chemical, biological, biotechnology, or radiopharmaceutical substance that has not been previously available for therapeutic use in humans and that is destined to be made available as a “prescription only medicine” to be used for the cure, alleviation, treatment, prevention or in vivo diagnosis of diseases in humans. The term NAS also includes:
- An isomer, mixture of isomers, or a complex or derivative or salt of a chemical substance previously available as a medicinal product but differing in properties with regard to safety and efficacy from that substance previously available.
- A biological or biotech substance previously available as a medicinal product but differing in molecular structure, nature of source material, or manufacturing process and which will require clinical investigation.
- A radiopharmaceutical substance that is a radionuclide or a ligand not previously available as a medicinal product. Alternatively, the coupling mechanism linking the molecule and the radionuclide has not been previously available.
- A combination that contains an NAS, even if it also contains another previously approved substance.
Applications that are excluded from the study:
- Any other application, where new clinical data were submitted.
- Generic applications.
- Those applications where a completely new dossier was submitted from a new company for the same indications as already approved for another company.
- Applications for a new or additional name, or a change of name, for an existing compound (i.e., a “cloned” application).
Data Sources
Data were collected from public assessment reports from the agency websites.
For EMA
For FDA
- https://www.fda.gov/vaccines-blood-biologics/licensed-biologicalproducts-supporting-documents. For EMA and FDA—specific to discordance of outcomes for orphan NAS approved by FDA and EMA:
The rationale for non-approval of certain NASs in one agency but not the other was extracted from the public domain, such as from agency websites, pharmaceutical company websites, and news articles.
Data Collection Process
For CIRS proprietary databases, a review of the product inclusion against the NAS definition as well as data collection was performed by three CIRS researchers. One researcher extracted the data, and a second researcher validated the data through an independent review. Discrepancies were discussed until consensus was reached, and the third researcher facilitated adjudication of any differences.
For additional information collected by CIRS for the purpose of this project:
- Alternative and confirmatory data: Based on a working definition and list of keywords provided by National Academies staff,4 two CIRS researchers worked on developing a method that was reviewed and adjudicated by a third CIRS researcher. Following, agreement on the method, which was also reviewed by the National Academies staff, one researcher collected the information, and the second researcher validated a subset of applications to ensure consistency of the method applied. Disagreements were adjudicated by a third CIRS researcher to reach agreement.
- Discordance: The method was developed by three CIRS researchers and reviewed with National Academies. Data collection was undertaken by one researcher and reviewed by the second. Disagreements were adjudicated by a third CIRS researcher to reach agreement.
Collected Characteristics
In addition to the brand name, generic name, and sponsor (applicant), the collected variables are outlined in Table D-1.
TABLE D-1
Variables and Data Points Collected for Each New Active Substance.
Year Ranges
The focus of the analysis was on NASs approved between January 1, 2013, and December 31, 2022.
Caveat:
- The collection of alternative and confirmatory data was limited to the following year ranges: 2013–2014, 2017–2018, 2021–2022. The sample was selected to manage the volume of data collection required, while still providing an overview of the decade.
- Discordance on the approval of orphan NAS between agencies: this analysis investigated regulatory review outcomes for orphan NASs approved by either FDA or EMA from 2018 through 2022; however, regulatory outcomes were tracked (at the other agency) into 2024 (public domain last accessed in April 2024).
Analysis of Time
For timelines, medians and percentiles (25th and 75th percentiles) were analyzed to facilitate the understanding of the variation around the median (50th percentile). The following time periods were calculated:
- Approval time: Time calculated from sponsor submission date to regulatory approval date. This time includes agency and company time. EMA time includes European Commission time.
- Submission gap: Date of submission at the first regulatory agency to the date of regulatory submission to the subsequent regulatory agency.
- Rollout time: Time between the date of submission at the first regulatory agency to the date of approval by the target agency.
Caveat: All timelines were calculated in calendar days (hereafter “days”).
REFERENCES
- Hirako M, McAuslane N, Salek S, Anderson C, Walker S. A comparison of the drug review process at five international regulatory agencies. Therapeutic Innovation & Regulatory Science. 2007;41:291–308.
- CIRS (Centre for Innovation in Regulatory Science). R&D Briefing 88: New drug approvals in six major authorities 2013–2022: Focus on orphan designation and facilitated regulatory pathways. London: Centre for Innovation in Regulatory Science; 2023. [July 12, 2024]. https://cirsci
.org/publications /cirs-rd-briefing-88-new-drug-approvals-in-six-major-authorities-2013-2022-focus-on-orphan-designation-and-facilitated-regulatory-pathways/
Footnotes
- 1
For more information https://www
.nationalacademies .org/our-work /processes-to-evaluate-the-safety-and-efficacy-of-drugs-for-rare-diseases-or-conditions-in-the-united-states-and-the-europeanunion (accessed December 11, 2023). - 2
A new active substance (NAS) was defined as a chemical, biological, biotechnology, or radiopharmaceutical substance that has not been previously available for therapeutic use in humans and is destined to be made available as a “prescription-only medicine” to be used for the cure, alleviation, treatment, prevention, or in vivo diagnosis of diseases in humans.
- 3
For FDA, it should be noted that certain pending applications are included (CR), whereas for EMA applications were included only where a final opinion was given (i.e., all submissions had a regulatory outcome).
- 4
For purposes of this data analysis, “alternative and confirmatory data” refers to marketing authorization data submitted to FDA or EMA that falls outside of an adequate and well controlled trial and may have been used by a given regulatory agency to evaluate safety or effectiveness of a drug product. Sources of supplemental data may include:
- Natural history studies (e.g., patient registries)
- Expanded-access programs
- Open-label extension studies
- External control groups (concurrent and historical)
- Case reports
- Extrapolation based on data from related drug products or indications
- Mechanistic correlation (pharmacokinetic and pharmacodynamic data)
- Nonclinical studies (e.g., stability and quality control data)
- Passive data collection (e.g., digital phenotyping)
- Patient and caregiver reported outcomes (including preference data)
- Real world evidence
- Literature reviews
- Centre for Innovation in Regulatory Science Data Analysis Methodology - Regulato...Centre for Innovation in Regulatory Science Data Analysis Methodology - Regulatory Processes for Rare Disease Drugs in the United States and European Union
Your browsing activity is empty.
Activity recording is turned off.
See more...