

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Committee on the Return of Individual-Specific Research Results Generated in Research Laboratories; Downey AS, Busta ER, Mancher M, et al., editors. Returning Individual Research Results to Participants: Guidance for a New Research Paradigm. Washington (DC): National Academies Press (US); 2018 Jul 10.

Returning Individual Research Results to Participants: Guidance for a New Research Paradigm.
Show detailsIn the preceding chapters, the committee has provided strategies that it envisions will, over time, result in improved access to individual results for those who have participated in research. At present, however, a number of legal and regulatory barriers impede the return of individual research results to participants and contribute to unevenness in access across states and research institutions. This chapter examines whether the regulatory environment is appropriately calibrated to the risks and benefits of participant access to research results and describes changes in the legal and regulatory landscape that are needed if the committee's recommendations are to be implemented and its vision for the return of results to participants as a more commonplace practice in research is to be achieved.
LEGAL AND REGULATORY PROTECTIONS IN RELATION TO THE BENEFITS AND RISKS OF RETURNING INDIVIDUAL RESEARCH RESULTS1
The legal and regulatory landscape pertaining to the return of individual research results is governed by a complex assortment of federal and state laws (including statutes and regulations, see Box 6-1). Currently in the United States, no federal law confers a fundamental right to access research results generated from biospecimens collected during the course of research,2 despite the perception among some individuals that they continue to own their biospecimens and any personal data generated from them (Obama, 2016; Rothstein, 2015). At least one scholar has argued that patients' and consumers' right of access to health-related data is a federal civil right provided by the Health Insurance Portability and Accountability Act of 1996 (HIPAA) Privacy Rule (Evans, 2018a), but there is no legal consensus or formal precedent about the breadth of this right.3 A few states (e.g., Colorado,4 Alaska5) have enacted statutes that recognize individuals' property rights to certain biological samples or the results of analyses on those specimens, or both, but in general state courts have held that biospecimens donated during research and the results of tests performed on those specimens are not the legal property of research participants (Francis, 2014).
BOX 6-1
Overview of Federal Laws and Regulations Relevant to the Return of Individual Research Results.
Despite this lack of legal consensus, a number of ethical and practical reasons for disclosing individual results (as discussed in Chapter 2) collectively form a strong argument for creating pathways to enable research participants access to their information. Indeed, the past few years have seen a significant shift in the regulatory environment toward allowing individuals greater access to their laboratory results. For example, since 2000 the Health Insurance Portability and Accountability Act (HIPAA) regulations have recognized the right of individuals to inspect and obtain a copy of their protected health information (PHI)6 contained within a designated record set (DRS).7 This right of access is binding on all HIPAA-covered entities,8 except that before 2014 this right did not apply to HIPAA-covered laboratories. Similarly, the original access rule for the Clinical Laboratory Improvement Amendments of 1988 (CLIA) did not allow the release of laboratory test results to the individual undergoing testing except in states that provided for direct access. Individuals not residing in these states were dependent on their ordering health care provider to share test results. But in 2014, in a final rule to CLIA and HIPAA,9 the Department of Health and Human Services (HHS) announced the elimination of the laboratory exclusion from the HIPAA access rule as well as the CLIA prohibition on the return of test results to individuals (Barnes et al., 2015; Federal Register, 2014). Although the change was primarily aimed at ensuring patient access to clinical laboratory information, the revised HIPAA access rule, which preempts any state laws that restrict an individual's direct access to test results, has opened doors for participants to access their individual research results. As currently written and implemented, however, the laws and regulations governing access to laboratory results are not harmonized; they afford inconsistent and inequitable access for participants to permitted results, and the regulatory conflicts create dilemmas for laboratories, forcing them to choose which regulation to intentionally violate in order to comply with the other. These issues and some approaches to their resolution are discussed below.
CLIA and Its Restrictions on Returning Individual Results
Laboratories in the United States that perform tests on human specimens for the purpose of providing information for the diagnosis, prevention, or treatment of a disease or for the assessment of the health of an individual are regulated by CLIA10 and are required by the Centers for Medicare & Medicaid Services (CMS) to be CLIA certified through a process that ensures that certain quality control assurances and requirements are in place (see Chapter 3). CLIA and its associated regulations were put in place to protect patients from harm resulting from inaccurate laboratory testing. Laboratories licensed in states that have enacted laws with requirements equal to or more stringent than those required under CLIA and where CMS has approved the licensure program qualify as CLIA exempt.11,12 CLIA also allows for an exception from certification requirements in the case of laboratories that conduct tests on human specimens for research purposes and that do not report patient-specific results for the diagnosis, prevention, or treatment of a disease or for the assessment of the health of an individual (CMS, 2014). If laboratories report individual results that could be used for clinical decision making, even if this is not the intended purpose of returning results, CMS has interpreted the regulations to mean that those laboratories must be CLIA certified. These laboratory categories are summarized in Table 6-1.
TABLE 6-1
CLIA Categories of Laboratories.
Many laboratories that generate research results with the potential for use in health care are CLIA certified. For example, CLIA-certified clinical laboratories may be (but are not always)13 used to conduct testing on biospecimens collected for clinical trials (Barnes et al., 2015). In such cases, CLIA does not pose any impediment to the return of research results (or to the return of clinical test results generated in the course of a research study). However, a number of potential scenarios in which results are clinically actionable or otherwise valuable to participants could be generated in laboratories that are not CLIA certified (e.g., academic or industry laboratories conducting more basic research, such as biomarker identification, using innovative methodologies that have not yet been validated for clinical use, or laboratories engaged in assay development). Research testing performed in laboratories that are not CLIA certified may create a dilemma for investigators and institutions that feel an obligation to return such results, particularly when they are urgent and might not otherwise be discovered. Under CMS's interpretation of CLIA, investigators would be prohibited from disclosing the results without first becoming CLIA certified. It has been argued that investigators may have a First Amendment right under the U.S. Constitution to share the results of genetic research tests with interested participants as a form of protected free speech (Evans, 2014). However, the validity of this argument has not yet been tested in court (Jarvik et al., 2014), and the potential consequences for CLIA violations may deter investigators from returning results generated in a non-CLIA-certified laboratory. CMS is authorized to impose a civil monetary penalty ($50–$10,000 per day of noncompliance per violation) and can file a civil lawsuit to obtain a court order that prohibits a laboratory from continuing an activity that CMS believes to represent a “significant hazard to the public health.”14 Although the committee is not aware of any cases where such enforcement actions have been taken against non-CLIA-certified laboratories for the return of research results, CMS recently intervened in the activities of a direct-to-consumer genetic testing firm, directing the company to obtain CLIA certification before providing genetic testing results to consumers (Lee, 2017).
The committee recognizes the importance of ensuring the quality and integrity of laboratory test results. If investigators and their research laboratories plan to offer individual research results to participants, the testing of human biospecimens under an acceptable quality management system is essential (see Recommendation 3). However, as discussed in Chapter 3, expecting all research laboratories to meet CLIA requirements in order to return research results may not be reasonable. The current absolute prohibition on the return of research results from non-CLIA-certified laboratories also fails to account for several factors, including the high quality maintained by some research laboratories, the value that many participants place on results despite uncertain validity, and the access rights afforded by HIPAA to individual results regardless of quality standards. Additionally, there is a paucity of evidence of harm from the return of research results, though as discussed in Chapter 2, the overall body of evidence is limited and “the current state of knowledge reflects lack of evidence, not evidence of a lack of effect.” Thus, as stated in Recommendation 11, it is important for investigators to expand the body of evidence on the benefits and harms associated with the practice as it evolves and becomes more widespread.
CONCLUSION: As many research laboratories are not CLIA certified, CLIA represents a formidable obstacle to the return of individual results to research participants even though the results may meet other quality standards and a right of access to laboratory results has been gaining credibility in other regulatory policies.
Concerns Regarding an Access Right to Research Results Under HIPAA
Since 2000, HIPAA has included a rule that individuals have a right of access to inspect and obtain a copy of their PHI maintained in a designated record set. According to guidance issued in 2002, “any research records or results that are actually maintained by the covered entity as part of a designated record set would be accessible to research participants unless one of the Privacy Rule's permitted exceptions applies” (OCR, 2013b). Prior to 2014, there was an explicit exception from the access right for all HIPAA-covered laboratories “subject to” CLIA and laboratories “exempt from” CLIA.15 As discussed earlier in this chapter, the 2014 amendment removed both of these exceptions.16 As a result of this amendment, all laboratories that are part of a covered entity, regardless of their status under CLIA, must now comply with the access right under the HIPAA Privacy Rule.17 This means that all of an individual's protected health information in the designated record set, regardless of whether it is generated in a clinical laboratory or during the course of research, must be provided to that individual upon request.
This recent regulatory change has been met with mixed reactions and has been a focus of significant controversy. The change has been welcomed by some as a critical step forward in the movement to acknowledge participants' critical contributions to research and to respect their desires to receive information about themselves, even research information. At the same time, however, the increased access has generated concern among some researchers, clinical care providers, and regulators by providing an access right to research results and even uninterpreted data (e.g., genomic sequence data), which may not meet the high standards for quality that are required of clinical test results (Barnes et al., 2015; Evans, 2018a). Laboratories may now be compelled to share research results with unclear meaning (e.g., genomic variants of unknown significance) or questionable validity, raising fears that, without proper context or a clear understanding of the limitations, the receipt of such results could cause undue anxiety or prompt unnecessary intervention. Institutions have also expressed concerns regarding the potential financial impacts of the disclosure requirements on laboratories, because the HIPAA access right is an unfunded federal mandate (Evans, 2018b).
In addition to these concerns, the expanded right of individuals to access their laboratory results under HIPAA creates the potential for conflict with other federal regulations related to the protection of patients and research participants, as discussed below.
HIPAA and CLIA
For those non-CLIA-certified research laboratories that are HIPAA-covered entities, a legal dilemma can arise if a participant requests individual research results that constitute PHI contained within a DRS (the respective obligations of laboratories regarding individual access to laboratory results under CLIA and HIPAA are summarized in Table 6-2). Returning research results that are part of the DRS in order to comply with the expanded HIPAA access rule would force non-CLIA-certified laboratories to either violate CLIA (as currently interpreted by CMS) or forfeit the exception for research laboratories and obtain CLIA certification, which has significant cost and resource implications (Barnes et al., 2015). One researcher who spoke with the committee during its public workshop had compared costs for a large-scale exome sequencing study using CLIA- and non-CLIA-certified laboratories and estimated a three-fold difference in cost for the data generation alone.18 As discussed in Chapter 3, opportunity costs associated with CLIA certification also pose barriers for many laboratories. HIPAA Privacy Rule violations, which are enforced by the HHS Office for Civil Rights, can be costly, with monetary penalties as high as $50,000 for each day that the covered entity is in violation (Barnes et al., 2015), putting laboratories without the means to obtain and maintain CLIA certification in the difficult position of having to weigh the relative risks of violating CLIA or HIPAA.
TABLE 6-2
Legal Obligations Related to Individual Access to Laboratory Test Results.
To avoid such conflicts, and in the absence of additional guidance from OCR, some research institutions are interpreting the definition of a designated record set to exclude research results and setting policies accordingly. The rationale is presumably that research results are often not used to make decisions about individuals, which is required in order for it to be considered part of the DRS. Johns Hopkins Medicine, for example, has indicated that, based on its interpretation, a research record is not part of the DRS and only information that is entered into a patient's medical record during research would be part of the DRS and therefore subject to the HIPAA access rule (Johns Hopkins Medicine, 2015). Similarly, New York University's Langone Health System has interpreted the definition of the DRS as excluding research results generated in laboratories not certified by CLIA. This approach, which creates variation across institutions in the ability of research participants to access their results, has been facilitated by ambiguity in the definition of the designated record set and a lack of clear guidance from OCR. The Secretary's Advisory Committee on Human Research Protections (SACHRP, 2015) and other groups have called for clarification of the duties of HIPAA-covered entities to provide results from laboratories that are not CLIA certified and for guidance on how the term DRS should be interpreted.
CONCLUSION: The operationalization of HIPAA access rights to include CLIA-exempt laboratories and those operating under the CLIA research exception creates an insoluble conflict between patients' right to access their research results contained within the DRS and non-CLIA-certified laboratories' prohibition from returning such results under the current CMS interpretation of CLIA.
CONCLUSION: OCR has contributed to the confusion of investigators by failing to define the DRS and, specifically, the status of results generated in research laboratories that are not CLIA certified.
HIPAA and the Common Rule
The Common Rule, codified in separate regulations by 15 federal departments and agencies, is federal policy for the protection of human participants in any research that is conducted, funded, supported, or otherwise subject to regulation by the federal government. The Common Rule outlines the basic provisions for institutional review boards (IRBs), informed consent, and assurances of compliance.19 Under the Common Rule, IRBs are required to review and approve study protocols involving human participants to ensure that the risks are reasonable relative to the anticipated benefits and that participation is conditioned on the informed consent of research participants. The Common Rule neither explicitly encourages nor explicitly prohibits the return of results to study participants, but pending revisions to the regulation will require investigators to disclose their plans for returning individual research results (i.e., whether results will be returned to participants and, if so, under what conditions). If a research laboratory is (or is part of) a covered entity, participants may be told that their results will not be offered to them, as required by the revisions, even though they will retain a right to access the results under HIPAA if the results are part of the DRS. As discussed in Chapter 5, in such cases participants should be informed of their HIPAA access rights during the informed consent process.
Investigational Device Exemption Regulations and Return of Individual Research Results
FDA regulates, among other things, devices “intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals.”20 FDA's investigational device exemption (IDE) offers a regulatory pathway for investigators using an investigational device that has not yet been approved or cleared by FDA for the purposes for which it will be used in a particular study. FDA broadly defines an investigational device as one “that is the object of an investigation”21 conducted for purposes of determining safety or effectiveness and involving one or more participants (Federal Register, 1980).
FDA's IDE process is intended to protect the interests of participants whose clinical care might be affected by the results of investigational devices. If a study proposes to use a laboratory test in a manner that has not been cleared or approved by FDA, the investigators, with oversight from their IRB, must determine whether the IDE regulations apply because the test is an investigational device. If so, investigators must next consider whether the study nevertheless qualifies as exempt from IDE regulations. An investigational device that is subject to the IDE regulations is exempt if it is non-invasive and not used for diagnostic purposes without confirmation of the diagnosis by a “medically established” diagnostic product or procedure. If the study does not qualify as exempt, the investigator must determine whether the device poses “significant risk” (i.e., presents a potential for serious risk to the health, safety, or welfare of a participant)22 or “non-significant risk.” Figure 6-1 shows this decision-making process and the implications for IDE requirements.
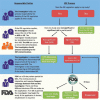
FIGURE 6-1
Investigational device exemption process for research studies. NOTE: FDA = Food and Drug Administration; IDE = investigational device exemption; IRB = institutional review board; NSR = non-significant risk; SR = significant risk. SOURCE: NHGRI, 2017.
The return of research results to research participants may affect the IDE process in at least two ways. First, in the case of studies involving devices, such as whole-genome sequencing, that are being used for purposes not yet cleared or approved by FDA, it appears that the return of results may play a role in determining whether the IDE regulations apply. A National Human Genome Research Institute (NHGRI) webpage focused on IDE regulations states,
If the investigator does not propose to return results to participants or their physicians, and the results will not otherwise be used to direct or inform the clinical care of that participant, then the investigational device study is exempt from the IDE regulation. (NHGRI, 2017)
Second, the return of research results to research participants may affect the risk classification of a non-exempt investigational device study. This risk classification determines the rigorousness of the requirements necessary to obtain an IDE. An abbreviated process can be used to obtain an IDE for devices that are not significant risk. In determining whether a device poses a significant risk, FDA considers many factors, such as the health status of the population under study and how the information generated by the device will be used in the study, including whether and how results will be returned. In a June 2016 workshop hosted by NHGRI, representatives from FDA explained that the risk of returning investigational device results to healthy volunteers would be considered different from the risks if members of the participant population have a disease or health condition (NHGRI, 2017).23 The NHGRI resource that reflects information learned from that workshop and the experience of NHGRI grantees describes an example of a significant risk study as one that
might involve genome sequencing of healthy participants with an intent to return variants of unknown significance (VUS). In this instance, the risk might stem from concern that test results with unknown clinical significance would lead healthy individuals to pursue unnecessary treatments that could expose them to harm. If this study design does not also include appropriate risk-mitigating measures, it could be considered SR [significant risk]. (NHGRI, 2017)
The committee is aware of only one reported case, the Newborn Sequencing in Genomic Medicine and Public Health program—North Carolina Study, in which the return of research results to healthy volunteers was a key consideration in FDA's determination that investigators needed to pursue a full IDE (NHGRI, 2016). However, the potential for the return of results to trigger additional regulatory hurdles is an additional barrier for investigators considering returning research results and may incentivize them against return.
FDA has not provided binding guidance on the role of the return of individual research results in the IDE process, including the determination of whether a device is an investigational device or the determination of whether the device poses significant risk. As a result, it is unclear how the IDE regulations actually affect most studies that return results in practice. It is also unclear if and how these analyses will change if investigators return results only in response to individual requests under HIPAA.
CONCLUSION: FDA's interpretation of how the return of individual research results triggers application of the IDE process and affects the risk assessment is unclear. FDA has introduced further confusion by suggesting that the return of individual research results for healthy individuals triggers a significant risk determination, requiring a full IDE.
CONCLUSION: It is unclear how access to individual research results under HIPAA affects FDA's risk assessment for IDEs.
Inconsistent Use of Terminology in Federal Regulations
Terminology that is inconsistent across different regulations can cause unnecessary ambiguity and confusion. The inconsistencies often relate to the process by which laws are written—i.e., different terms with origins in different laws may have the same meaning. In addition to the confusion created by different uses of the term “CLIA exempt,” as mentioned above, at least two other examples of terminology are ambiguous or confusing and need to be harmonized across the relevant regulations.
The first relates to terms used to describe the information that individuals can access under CLIA and HIPAA. The original CLIA access rule provided authorized persons access to “test results” generated in CLIA-regulated and CLIA-exempt laboratories. Of note, “authorized persons” did not include the patients themselves (or their personal representative) except in states that explicitly permitted direct patient access. Under the 2014 CLIA access rule, patients and their personal representatives can now access their “completed test reports.” The term “completed test report” was not defined, but the preamble to the access rule indicated that test results were complete when “all results associated with an ordered test are finalized and ready for release” (Federal Register, 2014, p. 7295). However, how the completed test report differs from the test results that can be accessed by the individuals authorized to order or receive them remains unclear. This ambiguity also contributes to confusion regarding research results that individuals can obtain under the HIPAA access rule, which allows individuals to access their protected health information that is contained within the DRS. The 2014 amendment to the HIPAA Privacy Rule states (in response to a comment) that laboratory test reports do not become part of the designated record set until they are complete.24 Some legal scholars have suggested that OCR could clarify that laboratory research results with uncertain clinical significance would not be considered “complete” and thus excluded from the DRS or, alternatively, could issue guidance that research test results are explicitly included in the DRS regardless of their clinical significance (Barnes et al., 2015).
The second terminology issue relates to the description of the identifiability of results. Federal regulations use different terms based on the standards applied when removing the linking information. According to HHS convention, “de-identified” describes results where the linking information has been removed in accordance with HIPAA standards,25 while “non-identified” refers to results where the linking information has been removed in accordance with the Common Rule, which prescribes standards that are different than HIPAA standards (OHRP, 2016a).26 Adding further confusion is the use of the term “identifiable sensitive information” in the 21st Century Cures Act in reference to certificates of confidentiality.
CONCLUSION: Inconsistent use of terminology by different regulatory agencies creates unnecessary ambiguity and confusion for investigators, institutions, and research participants.
Discrimination Concerns for Research Participants
The possibility that participants who receive individual research results may face discrimination if the results indicate a previously undiagnosed condition (or a genetic predisposition for a condition) is a concern among both proponents and opponents of the practice. Federal and state laws provide some protections for individuals against discrimination on the basis of disabilities, pre-existing conditions, and genetic information, but gaps remain.
First, the Americans with Disabilities Act (ADA) prohibits discrimination against individuals with disabilities in the areas of employment, public services, and public accommodations.27 Specifically, employers are prohibited from “discriminating against individuals who (with or without reasonable accommodation) can perform the essential functions of employment positions in hiring, promotion, training, compensation, and other job-related decisions.”28 Additionally, employers cannot conduct medical examinations or ask questions of job applicants and employees regarding whether they have a disability or the nature or severity of their disability unless the questions are related to the applicant or employee's job.29 However, employers may require individuals to release their health records after making a conditional offer of employment.
The Genetic Information Nondiscrimination Act of 2008 (GINA)30 prohibits discrimination in health insurance and employment contexts based on genetic information, which is defined as information about an individual's genetic tests, about the genetic tests of family members, or about the presence of a disease or disorder in family members. Congress's intent in passing GINA was to remove barriers to genetic testing for personal health and research purposes by providing legal protection against the misuse of genetic test results. GINA expanded protections instituted earlier through HIPAA by prohibiting group and individual insurers from using genetic information to set premiums or make eligibility decisions for individuals or groups. The law further prohibits health plans from requesting or requiring genetic testing prior to an individual's insurance enrollment and from requesting, requiring, or purchasing genetic information for underwriting purposes.31
GINA similarly limits both access to and use of genetic information by employers (Prince and Berkman, 2012).32 Thus, employers are prohibited from requesting, requiring, or purchasing genetic information about employees or their family members.33 Employers also may not use genetic information to make employment decisions (e.g., hiring, firing, promotion, and compensation) or to deprive employees of employment opportunities.34 Finally, employers must treat the genetic information of their employees as confidential medical records that generally may not be disclosed.35
While GINA affords important protections for patients and research participants, it notably does not regulate discrimination based on non-genetic information (e.g., a positive test result for a protein biomarker for Alzheimer's disease) (Arias and Karlawish, 2014). Moreover, GINA is limited to genetic information and does not apply to discrimination based on already expressed genetic conditions, although the Patient Protection and Affordable Care Act closed this loophole in health insurance by prohibiting individual and group insurers from denying coverage based on any kind of pre-existing condition.36 GINA also does not confer protection against the discriminatory use of genetic information by life, disability, or long-term care insurers or in other contexts in which discrimination may occur, such as housing and education. These gaps in GINA's protections may discourage people from participating in biomedical research, particularly when there is the potential to uncover unanticipated findings indicating an increased risk for a health condition (Green et al., 2015).
Most states have sought to augment federal anti-discrimination protections related to genetic information. In a few cases, state laws have focused on preventing discrimination against individuals with specific genetic traits (e.g., sickle cell trait or cystic fibrosis trait), but the majority are more broadly aimed at preventing the discriminatory use of genetic information in employment and insurance decisions. Twenty-four states have enacted laws addressing gaps in GINA's protections by limiting genetic discrimination in life, disability, or long-term care insurance, and California's law further bars discrimination in emergency medical services, housing, mortgage lending, and public education. The implication of this patchwork of statutes is that the protections for research participants who choose to receive their research results will vary by geographic location, and participants will need to understand the anti-discrimination protections in their state.
CONCLUSION: Research participants who choose to receive results from the testing of their biospecimens (or who exercise their right of access under HIPAA) should be informed about the discrimination protections under GINA and the ADA and relevant state laws and what GINA/ADA/state laws do not protect against.
Liability Concerns for Investigators, Laboratories, and Institutions
Survey data have indicated that a fear of legal liability influences investigators' decisions on whether to return individual research results (Ramoni et al., 2013). Concerns regarding the liability associated with not returning results can motivate some investigators to disclose research results to participants, but, at the same time, fears of lawsuits stemming from inaccurate findings or medical mismanagement subsequent to the receipt of results may discourage investigators from returning them.
The legal liability for investigators concerning the return of individual research results most likely appears in the form of tort liability,37 which falls under state law. In the absence of federal regulations or case law directly addressing a researcher's duty to disclose significant research results, investigators and their institutions are left to make difficult decisions about whether and when to return results in the context of significant legal uncertainty surrounding this issue (McGuire et al., 2014).
Concerns Regarding Liability for Disclosure
Several circumstances associated with the return of individual research results may give rise to tort liability. These include
- the disclosure of correct results (of timely interest or utility) to the wrong individual, which might be caused by the improper labeling of biospecimens or results, especially in laboratories without a quality system (this situation could also create liability for non-disclosure—discussed in the next section—if the results are not disclosed to the right individual);
- the disclosure of incorrect results to the right individual, which might be caused by a mishandling of biospecimens, improper test administration, or misinterpretation (e.g., negligent misclassification of variants);
- the disclosure of results to individuals who are not authorized to receive them; and
- failure to update previously disclosed results and to return the updated results.
Although research laboratories are not subject to the same quality requirements as clinical laboratories, they still must maximize the analytic and clinical validity of any results returned to participants. For example, if investigators return erroneous results generated by a research laboratory without indicating that the results need to be verified in a CLIA-certified laboratory before any clinical actions are undertaken, the researchers may be liable to a tort filing. However, even in the case that appropriate warnings regarding the need for confirmatory clinical testing were provided, it is not clear whether this would be sufficient to protect investigators from tort liability (McGuire et al., 2014). This will be decided by courts on a case-by-case basis. For rapidly evolving fields like genomics, further complications arise from continuing changes in the standard of care regarding interpretation and the return of results.
Concerns Regarding Liability for Non-Disclosure
Tort liability related to non-disclosure is most likely to arise as a form of negligence, where an individual owed a duty to another person but breached that duty, and the person was harmed as a result (McGuire et al., 2014). In the return-of-results context, this might involve a case where an investigator or laboratory failed to return urgent, medically actionable results and the participant was harmed as a result.
Whether one person owes a legal duty to another depends on the nature of their relationship and is highly context specific (Pike et al., 2014). As an example, fiduciary relationships—two-way relationships based on trust—can give rise to a claim of negligence when the fiduciary does not act in the best interests of the principal. However, except in cases where investigators are also a participant's treating physician, the courts have declined to view researchers as fiduciaries of research participants.38 Nevertheless, precedent suggests that researchers may have a “special relationship” to participants that gives rise to a duty to disclose certain research results (see the Grimes v. Kennedy Krieger Institute case described in Appendix C, p. 330, Special relationships).
Whether a special relationship between investigators and participants gives rise to duties that include the return of certain individual research results also depends on the prevailing standard of care. The standard of care can be established by guidance and recommendations to return results, by recognition by scholars and the research community of an ethical obligation to return results, or by a common practice of returning results. Consequently, if the practice of returning results becomes routine, legal scholars have noted, researchers will be legally required to do so (Clayton and McGuire, 2012). Thus far, however, ethics-based recommendations from expert groups have not yet been used to impose legal liability (Wolf, 2012).
Given the concerns regarding legal liability, a staged approach to expanding the practice of returning individual research results (discussed in Chapter 4) may generate forward momentum while allowing the field to test the legal waters and work out the early precedents. As stated by one legal scholar, “to shape ethics and practice around premature conclusions of legal threat would be to thwart an extremely important debate in research ethics and practice” (Wolf, 2012).
CONCLUSION: Perceived liability risks may dampen interest in returning individual results to research participants. However, good faith efforts by investigators to return individual research results with proper warnings and caveats are unlikely to result in legal action for negligence. Delineating standards for the return of individual research results will help to mitigate concerns regarding liability.
CREATING A REGULATORY ENVIRONMENT BETTER ALIGNED WITH THE BENEFITS AND RISKS OF THE RETURN OF INDIVIDUAL RESEARCH RESULTS
The committee was asked to review the current regulatory requirements and consider whether current regulations are effective in minimizing the risks while maximizing the benefits of returning individual research results. In its assessment of the regulatory environment—described in the sections above—the committee found considerable confusion concerning the legal and regulatory requirements and restrictions pertaining to the return of individual results, causing variable interpretation and action across IRBs and research sites. This confusion stems, in part, from inconsistencies and ambiguities in the regulatory language as well as the potential for conflicts between federal regulations. In some cases, regulations or the institutional interpretations of the regulations are too restrictive, placing institutional protection ahead of participants' right to access health data, while others are not restrictive enough, allowing for the return of results of poor or unsubstantiated quality without appropriate disclaimers to inform participants regarding limitations in the validity and utility of the information they receive. These conflicts appear to result, in part, from opposing values of the federal agencies with oversight in this domain. The statutory responsibilities of CMS and FDA emphasize patient and participant safety, while the promotion of access is a core tenet of OCR's civil rights mandate. Recognizing the critical importance of both sets of values, the committee sees opportunity to strike a balance.
CONCLUSION: Overall the current regulatory environment for the return of individual research results is overly restrictive and characterized by confusion and conflict, unnecessarily impeding participants' access to research results that may have value to them.
CONCLUSION: Inconsistencies in terminology and discrepancies in requirements across different federal regulations relevant to the return of individual research results contribute to the confusion, but could be addressed through an effort to harmonize regulations.
In developing its recommendations on changes to better align the regulatory environment for the return of individual research results with the risks and benefits to individuals, the research enterprise, and society, the committee carefully considered the pros and cons of different approaches as well as the potential downstream effects on the current regulatory framework. The options presented in the sections below are not all mutually exclusive, and, in fact, a combination of approaches may be required to address different concerns.
Considerations for Changes to CLIA Regulations
CLIA regulations serve an important function in ensuring that the clinical results returned to patients are high quality and likely to be accurate. Requiring research laboratories to obtain CLIA certification before returning individual research results similarly increases the likelihood that the research results returned to participants are valid. However, the burdens associated with CLIA certification (e.g., direct costs, human resource requirements) as well as the inconsistencies between some of the CLIA regulations and the functions of a research laboratory create an incentive for investigators to plan not to return results, which, given participants' interest in receiving results, may adversely affect public participation in biomedical research (Crawley, 2001; Hiratsuka et al., 2012; Northington Gamble, 2006). Even in the absence of a plan to return results, investigators and decision makers in their institutions may face a moral dilemma in the event that an unanticipated result generated in a laboratory that is not CLIA certified has implications for the participant's well-being that are so great as to create an ethical imperative to return the result. Moreover, as described above, current conflicting regulatory requirements for CLIA and HIPAA can create legal dilemmas for laboratories that are covered entities, forcing them to choose which of the regulations to intentionally violate in order to comply with the other. Some have suggested that having a plan and allocating funds in the research budget for confirmatory testing in a CLIA-certified laboratory when a need arises to return unanticipated results could help resolve these kinds of dilemmas (Beskow and O'Rourke, 2015; Bookman et al., 2006). However, this assumes that additional samples can be collected from the participant, which is not always possible in research studies, and that a CLIA-certified laboratory is able to validate the research result. For some cutting-edge technologies and novel assays, a validated test in a CLIA-certified laboratory may not be available (Prucka et al., 2015). For example, if the research laboratory is performing a test for a new serum biomarker for a stroke, a CLIA-certified laboratory may not have a validated test for the biomarker. Even when retesting is possible, given the added cost of doing so, the committee does not believe that the possibility of retesting will address the current disincentives for returning research results to participants on a more routine basis. Therefore, the committee considers and describes below proposed changes to the CLIA regulations. In the absence of changes to the CLIA regulations,39 the committee believes that at the very least HIPAA regulations need to be changed or clarified so that non-CLIA-certified laboratories cannot be compelled to disclose PHI in the DRS of questionable analytic validity that are requested by participants. Potential changes to HIPAA regulations are discussed in the next section.
On one end of the range of options is a requirement that all testing on human biospecimens (not just those returning results) be conducted in CLIA-certified laboratories. In addition to removing current disincentives to return research results, resolving any possible CLIA–HIPAA conflict, and increasing confidence in the validity of results being returned to participants (both in planned and unplanned return scenarios), such a requirement could help address the broader concerns regarding the quality of research results and the reproducibility in biomedical research. However, the committee did not consider this a viable option, given the likely adverse impacts on many research laboratories, both in terms of the sustainability of costs (particularly for new and smaller laboratories operating earlier in the translational research continuum) and the impacts on innovation. A large number of laboratories conducting research on human biospecimens would require assistance (both financial and technical) for the staff training and operational changes that would be needed to obtain CLIA certification. Institutional core resources could help ameliorate this burden for individual laboratories/investigators, but not all institutions have the capacity to develop this kind of centralized infrastructure. Several groups have concluded that it would be infeasible for all research laboratories that test human biospecimens to become CLIA certified (Barnes et al., 2015; SACHRP, 2016) and such a requirement could disincentivize research on biospecimens, thereby slowing progress in some important research areas (e.g., assay development and novel biomarker identification).
On the other end of the continuum is the option for CMS to amend CLIA regulations so that research results could be returned to participants from any research laboratory without restriction (regardless of whether the laboratory operated under a quality management system and with no requirement for oversight). Although this would remove the current disincentive associated with the burden of CLIA certification for laboratories interested in returning research results, the committee rejected this option because of the potential harms associated with returning results lacking validity, particularly if the results are not accompanied by information conveying the limitations of the reliability and interpretation. The committee also was concerned that this would create a perverse incentive to test human biospecimens for clinical decision making in research laboratories in order to avoid the burden associated with meeting and maintaining the CLIA certification requirements.
Between these two extremes is a third option for CLIA regulatory changes which allows investigators and their institutions greater flexibility in determining the conditions under which returning results is appropriate when they have not been generated in a CLIA-certified laboratory. For the safety reasons discussed in Chapter 3, investigators planning to return research results that are intended for use in clinical decision making should ensure that those results are generated (or verified prior to use and return) in a CLIA-certified laboratory. However, the committee also recognizes that in some scenarios CLIA certification may not be appropriate to the nature of the research conducted by a laboratory and therefore recommends that the National Institutes of Health (NIH) lead the development of an externally accountable40 quality management system for research laboratories testing human biospecimens (see Recommendation 2).
The adoption of this standard by research laboratories would adequately support confidence in the analytic validity of research results so that they can be offered to participants, but it would require CMS to revise the CLIA regulations to allow for the return of results from laboratories operating under this research quality management system when the results are not intended for use in clinical decision making (see Box 3-1). To prevent abuse (i.e., by unscrupulous parties seeking to avoid CLIA certification costs and resources even when results are intended to inform clinical decision making) and protect the well-being of research participants, this exception from CLIA requirements would necessitate institutional processes (and the associated infrastructure and resources) for laboratory oversight and risk management. For example, external assessments of a laboratory's quality management system could be reported to the IRB as a condition of protocol approval prior to starting research incorporating the planned return of individual research results not intended for clinical decision making.
Prior to the establishment of an externally accountable quality management system for research laboratories or in cases where investigators did not plan to return results (i.e., unanticipated results or results requested by participants under HIPAA), the proposed CLIA exception could further allow decisions regarding the return of individual research results to be made at the institutional level contingent on an IRB review and approval process (see Recommendation 3). All research results returned to participants would need to be qualified with information that helps recipients understand the limits of the information being returned in order to minimize the risk of harm from a misinterpretation or misapplication of the results.
Considerations for Changes to the HIPAA Access Right
The proposed changes to the CLIA regulations to allow the return of individual research results from laboratories operating under the NIH-defined externally accountable quality management system or following an IRB review and approval process (discussed above and in Chapter 3) would go a long way toward resolving the CLIA–HIPAA conflict. However, it would still leave open the issue that, under the HIPAA access right, laboratories could be compelled to share research results with questionable validity or a high potential for misinterpretation. Some may argue that individuals should still have a right to access their information even if it is poor quality, and in regards to other types of information in the DRS, such as physician notes and electronic medical records (EMRs) information, that is the case. However, there is a crucial difference between laboratory results and these other types of protected health information—an individual has no way to know or verify if the laboratory result is accurate or if it even belongs to the individual. By contrast, a patient or patient's caregiver may recognize by himself or herself (or in consultation with the provider) that the information in the EMRs is incorrect. Thus, it is important to have some mechanism for individuals to have confidence in the quality of laboratory test results. Given the inherent uncertainty in research, results with these characteristics may be generated even when laboratories have established quality management systems in place, and investigators and institutions may not feel comfortable returning such results. Therefore, the committee considered additional regulatory changes related to the HIPAA right of access. While cautious about restricting one of the currently available pathways for participants to access their research results, particularly in an environment where results are rarely offered to participants, the committee believes that carefully considered changes are warranted to reduce the risk of harm from the return of research results.
HIPAA's access right applies only to laboratories that are—or are part of—a covered entity and to results that are maintained within the DRS for that entity. Consequently, two41 different pathways are proposed that restrict the right of access to research results: research laboratories can be legally separated from the HIPAA-covered part of the entity so that they are no longer subject to HIPAA's requirements, or the DRS can be defined to exclude all or some research results.
The legal mechanism for implementing the first approach is the creation of a hybrid entity (Barnes et al., 2015), which is an entity whose business functions include both covered and non-covered functions.42 Some academic medical centers have elected to become hybrid entities, with research laboratories that do not conduct covered transactions excluded from the covered portion of the entity. Such laboratories could not be compelled to provide access to research results under HIPAA.
However, a number of significant challenges and costs to creating a hybrid entity limit the viability of this approach on a wide scale (Barnes et al., 2015). First, some personnel may be involved with both covered (e.g., health care delivery) and non-covered (e.g., some research43) functions, making separation of the subparts difficult to define. The conduct of covered transactions by the non-covered part of the hybrid entity could result in a fine from the HHS OCR. For a fee, legal and other consultants can help institutions properly separate components into covered and non-covered elements. Second, in accordance with the HIPAA Privacy Rule, institutions would need to ensure that information from the covered part of the entity is shared with the non-covered part in the same way that information from any two separate entities is shared. This would necessitate separate information systems, which conflicts with the proposed use of the medical EHR for communication of research results noted above. Hybrid entities cannot have a unified electronic record system. In addition to the infrastructure costs associated with separate information systems, creating a hybrid entity restricts data sharing across covered and non-covered components44 (Barnes et al., 2015) and could thereby impede some kinds of research—e.g., research involving the review and analysis of medical records cannot be conducted by laboratories in the non-covered part of the entity. Moreover, while the creation of hybrid entities can be done on an institution-by-institution basis, no regulatory mechanism universally excludes research laboratories from the covered portion of an entity. Thus, while this approach may make sense for some individual institutions, the committee does not consider it to be a viable option for addressing the issues with HIPAA access to results of questionable validity and its potential conflict with the CLIA requirements.
An alternative approach that could have broader reach if implemented by OCR is to more explicitly define what is and is not included in the DRS. Defining the DRS so that it does not include research results—specifically those results not intended for clinical decision making45—would prevent laboratories from being compelled to disclose such results when requested by participants (although investigators and institutions could still decide to return the results if they were generated in a laboratory with an accepted externally accountable quality management system or if the disclosure was reviewed through an independent process and approved by the IRB per Recommendation 3). The committee notes that some research institutions are already taking this approach, but by each institution documenting its interpretation of what OCR intends to be included in the DRS in its own way, different standards are being created at different institutions, resulting in inconsistent and inequitable access to research results for participants.
Although defining the DRS to exclude all research results not intended for clinical decision making would resolve the issues of access to unreliable results and the potential CLIA–HIPAA conflict, this approach is unnecessarily restrictive since it also impedes access to high-quality research results that may have value to participants. Additionally, the fact that HIPAA excludes the right of access for protected health information only during the duration of the research study suggests that OCR did intend for research results to be part of the DRS once a study has reached completion. Instead, the committee favors a more inclusive approach to defining the DRS that would preserve access to individual research results generated in CLIA-certified laboratories and laboratories with a quality management system consistent with Recommendation 2 (see Figure 6-2). This approach reduces the risks (to participants and investigators) from returning unreliable results while maximizing access to reliable research results.
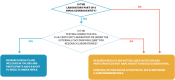
FIGURE 6-2
Determining whether participants have the right to access their individual research results under HIPAA. a CLIA-certified includes tests run in a CLIA-certified, -accredited, or -waived laboratory. b See Recommendation 2.
Recommendation 12: Revise and Harmonize Regulations to Support the Return of Individual Research Results.
Regulators and policy makers should revise and harmonize the relevant regulations in a way that respects the interests of research participants in obtaining individual research results and appropriately balances the competing considerations of safety, quality, and burdens on the research enterprise.
Specific actions that should be taken include
- A.
Because the designated record set (DRS) is intended to include information used to make decisions about individuals, those decisions should be based on test results that are of sufficient quality to be valuable for decision making. Accordingly, the Office for Civil Rights (OCR) of the Department of Health and Human Services (HHS) should define the DRS to include only individual research results generated in a CLIA-certified laboratory or under the externally accountable quality management system for research laboratories (see Recommendation 2);
- B.
OCR should require all HIPAA-covered entities that conduct research on human biospecimens to develop a plan that is reviewed and approved by the IRB for the release of individual research results in the designated record set to participants in a responsive manner when requested under HIPAA;
- C.
CMS should revise CLIA regulations such that when there is a legal obligation under the HIPAA access right to return individual research results, a laboratory will not be considered in violation of CLIA and need not obtain CLIA certification before satisfying this legal obligation;
- D.
CMS should revise CLIA regulations to allow research results to be returned from a non-CLIA-certified laboratory when they are not intended for clinical decision making in the study protocol (as defined in Box 3-1) and the laboratory conducts its testing under the quality management system with external accountability or the IRB has approved the return of results (as described in Recommendation 3);
- E.
CMS and OCR should harmonize the definitions of the following terms, providing a clear explanation and justification for any differences or discrepancies: “test report” and “completed test report” (CLIA), and “PHI in the designated record set” (HIPAA);
- F.
OCR, OHRP, and NIH should harmonize the definitions of the following terms, providing a clear explanation and justification for any differences or discrepancies: “de-identified” (HIPAA), “non-identified” (Common Rule), and “identifiable sensitive information” (21st Century Cures Act regarding certificates of confidentiality);
- G.
HHS (including CMS, FDA, NIH, OHRP) should ensure that all regulations, policies, and guidance relevant to human research refer to research “participants,” rather than research “subjects,” in accordance with the ethical principles of autonomy and respect for persons; and
- H.
FDA should clarify and provide additional guidance that if a device is not exempt from investigational device exemption (IDE) regulations, disclosure of results in many circumstances, including to healthy volunteers, will not necessarily entail significant risk, and FDA should clarify when it will consider the return of individual research results to entail significant risk. Additionally, FDA should provide guidance to IRBs on how to determine significant risk if the device is not exempt from IDE regulations.
FINAL THOUGHTS
The recommendations in this report, if followed, will result in substantial and potentially controversial changes to the research regulations and the research enterprise involving research with human biospecimens. The opportunity for change is due to the evolving relationship between investigators and participants, but more specifically to an assessment of the benefits and risk of results disclosure. The need for higher standards of quality in many research laboratories is clearly illustrated in this report and in our recommendations. Yet, despite the inherent limitations in the validity and reliability of many research results, our assessment is that the risks associated with the communication of results have been overstated, particularly for the many research projects that are unlikely to yield highly sensitive or clinically impactful results. Furthermore, the benefits to individual participants and to the research enterprise of results disclosure have been understated. Therefore, we are recommending that the absolute standard—that is, that all disclosed results must be generated in a CLIA-certified laboratory—should be replaced with a process-oriented standard, meaning that a peer-review process can be used in some circumstances to weigh competing considerations regarding the return of individual results. We recommend that such a process take into account, on a case-by-case basis, the values of the participants, the risks and benefits of the return of particular results, the quality of the research laboratory, and the feasibility for investigators to pursue this course. Moving away from an absolute standard has risks, but we believe that the risks can be mitigated through improvements in laboratory quality, a case-by-case assessment of the risks and benefits, and the promotion and development of communication strategies to help place results in the proper context for participants. The committee believes that the benefits of this more nuanced approach will greatly exceed the adverse impacts and costs.
The committee is well aware that more frequent return of individual research results will create new demands on the research and clinical enterprise. Many institutions and researchers currently lack the experience and resources to return individual research results in a deliberate and effective manner. The committee does not expect that the consistent and widespread return of individual research results will happen overnight. However, the committee foresees an evolving set of responsibilities and offers recommendations that it believes will help stakeholders prepare for these added responsibilities and develop the necessary expertise over time.
At a broader level, the justification for fundamental changes in the research landscape is found in our evolving understanding of the ethics of human participant research (which should be reflected in the language of federal regulations—see Recommendation 12G above) as well as to our recognition that failures to support transparency and to earn respect and trust from individuals in the community are hampering the conduct of science. The vision is that a dedicated commitment to collaboration will better honor participants, benefit science, and promote the welfare of society. The standards and practices related to the return of individual results are but one set of elements in this evolving landscape. But the return of individual research results is a tangible, measurable piece that we know is valued by participants and is feasible in many more circumstances than are reflected in current practice. Our hope is that this report will promote the return of individual research results through selected changes in research regulations, the use of quality systems that improve the quality of research results, and through the commitment of all stakeholders (see Table 6-3) to innovative, collaborative processes in the planning and conduct of research.
TABLE 6-3
Recommendations by Stakeholder.
REFERENCES
- Arias JJ, Karlawish J. Confidentiality in preclinical Alzheimer disease studies when research and medical records meet. Neurology. 2014;82(8):725–729. [PMC free article: PMC3945659] [PubMed: 24477112]
- Barnes M, Stayin S, Forster D, Russell-Einhorn M, Peloquin D, Medina-Jordan A. The CLIA/HIPAA conundrum of returning test results to research participants. Medical Research Law & Policy Report. 2015;14:491.
- Beskow LM, O'Rourke PP. Return of genetic research results to participants and families: IRB perspectives and roles. The Journal of Law, Medicine & Ethics. 2015;43(3):502–513. [PMC free article: PMC4617597] [PubMed: 26479559]
- Bookman EB, Langehorne AA, Eckfeldt JH, Glass KC, Jarvik GP, Klag M, Koski G, Motulsky A, Wilfond B, Manolio TA, Fabsitz RR, Luepker RV. Reporting genetic results in research studies: Summary and recommendations of an NHLBI working group. American Journal of Medical Genetics Part A. 2006;140(10):1033–1040. [PMC free article: PMC2556074] [PubMed: 16575896]
- Clayton EW, McGuire AL. The legal risks of returning results of genomics research. Genetics in Medicine. 2012;14(4):473–477. [PMC free article: PMC3779603] [PubMed: 22323070]
- CMS (Centers for Medicare & Medicaid Services). Clinical Laboratory Improvement Amendments (CLIA): How to obtain a CLIA certificate. 2006. [May 28, 2018]. https://www
.cms.gov/Regulations-and-Guidance /Legislation/CLIA /downloads/HowObtainCLIACertificate.pdf. - CMS. Research testing and Clinical Laboratory Improvement Amendments of 1988 (CLIA) regulations. 2014. [May 28, 2018]. https://www
.cms.gov/Regulations-and-Guidance /Legislation/CLIA/index .html?redirect=/CLIA. - CMS. List of exempt states under the Clinical Laboratory Improvement Amendments (CLIA). 2017. [May 28, 2018]. www
.cms.gov/Regulations-and-Guidance /Legislation /CLIA/Downloads /ExemptStatesList.pdf. - Crawley LM. African-American participation in clinical trials: Situating trust and trustworthiness. Journal of the National Medical Association. 2001;93(12 Suppl):14S–17S. [PMC free article: PMC2719992] [PubMed: 11798059]
- Evans BJ. The First Amendment right to speak about the human genome. Journal of Constitutional Law. 2014;16(3):549–636. [PMC free article: PMC4249692] [PubMed: 25473380]
- Evans BJ. William & Mary Law Review. Apr 1, 2018a. 2018. The Genetic Information Nondiscrimination Act at age 10: GINA's controversial assertion that data transparency protects privacy and civil rights. Forthcoming. Available at SSRN: https://ssrn
.com/abstract=3154009. [PMC free article: PMC8095822] [PubMed: 33953451] - Evans BJ. HIPAA's individual right of access to genomic data: Reconciling safety and civil rights. American Journal of Human Genetics. 2018b;102(1):5–10. [PMC free article: PMC5777935] [PubMed: 29304376]
- Ezzelle J, Rodriguez-Chavez IR, Darden JM, Stirewalt M, Kunwar N, Hitchcock R, Walter T, D'Souza MP. Guidelines on good clinical laboratory practice: Bridging operations between research and clinical research laboratories. Journal of Pharmaceutical and Biomedical Analysis. 2008;46(1):18–29. [PMC free article: PMC2213906] [PubMed: 18037599]
- Medical devices: Procedures for investigational device exemptions. Federal Register. 1980;45(13):3732–3759. Federal Register. [PubMed: 10248822]
- CLIA program and HIPAA Privacy Rule: Patients' access to test reports. Federal Register. 2014;79(25):7290–7316. Federal Register. [PubMed: 24605389]
- Francis LP. Genomic knowledge sharing: A review of the ethical and legal issues. Applied & Translational Genomics. 2014;3(4):111–115. [PMC free article: PMC4888827] [PubMed: 27294025]
- Green RC, Lautenbach D, McGuire AL. GINA, genetic discrimination, and genomic medicine. New England Journal of Medicine. 2015;372(5):397–399. [PubMed: 25629736]
- HHS (Department of Health and Human Services). Individuals' right under HIPAA to access their health information. 45 CFR § 164.524. 2016. [July 12, 2017]. https://www
.hhs.gov/hipaa /for-professionals /privacy/guidance/access/index.html. - Hiratsuka VY, Brown JK, Hoeft TJ, Dillard DA. Alaska native people's perceptions, understandings, and expectations for research involving biological specimens. International Journal of Circumpolar Health. 2012;71 [PMC free article: PMC3417706] [PubMed: 22663942] [CrossRef]
- Jarvik GP, Amendola LM, Berg JS, Brothers K, Clayton EW, Chung W, Evans BJ, Evans JP, Fullerton SM, Gallego CJ, Garrison NA, Gray SW, Holm IA, Kullo IJ, Lehmann LS, McCarty C, Prows CA, Rehm HL, Sharp RR, Salama J, Sanderson S, Van Driest SL, Williams MS, Wolf SM, Wolf WA, eMERGE Act-ROR Committee. CERC Committee. CSER Act-ROR Working Group. Burke W. Return of genomic results to research participants: The floor, the ceiling, and the choices in between. American Journal of Human Genetics. 2014;94(6):818–826. [PMC free article: PMC4121476] [PubMed: 24814192]
- Johns Hopkins Medicine. HIPAA questions and answers relating to research. 2015. [March 8, 2018]. https://www
.hopkinsmedicine .org/institutional_review_board /hipaa_research /faq_research.html. - Lee SM. BuzzFeed News. Nov 3, 2017. [March 8, 2018]. This DNA testing company is violating federal lab testing rules. https://www
.buzzfeed .com/stephaniemlee/orig3n-is-not-clia-certified-government-says?utm_term= .gyn5Go8Rd#.rtabzRp0e. - Legal Information Institute. Tort. 2018. [May 16, 2018]. https://www
.law.cornell.edu/wex/tort. - McGuire AL, Knoppers B, Zawati M, Clayton EW. Can I be sued for that? Liability risk and the disclosure of clinically significant genetic research findings. Genome Research. 2014;24(5):719–723. [PMC free article: PMC4009601] [PubMed: 24676095]
- NHGRI (National Human Genome Research Institute). National Human Genome Research Institute (NHGRI) investigational device exemptions (IDEs) and genomics workshop: Meeting report: Summary, standing questions, and next steps. Rockville, MD: National Human Genome Research Institute; 2016.
- NHGRI. Points to consider regarding the Food and Drug Administration's investigational device exemption regulations in the context of genomics research. 2017. [March 28, 2018]. https://www
.genome.gov /27561291/points-to-consider-in-assessing-when-an-investigational-device-exemption-ide-might-be-needed. - Northington Gamble V. Trust, medical care, and racial and ethinic minorities. In: Satcher D, Pamies RJ, Woelfl NN, editors. Multicultural medicine and health disparities. New York: McGraw-Hill; 2006. pp. 437–448.
- Obama B. The White House. 2016. [March 8, 2018]. Remarks by the president in precision medicine panel discussion. https:
//obamawhitehouse .archives.gov/the-press-office /2016/02 /25/remarks-president-precision-medicine-panel-discussion. - OCR (Office for Civil Rights). Summary of the HIPAA Privacy Rule. 2013a. [March 28, 2018]. https://www
.hhs.gov/hipaa /for-professionals /privacy/laws-regulations/index.html. - OCR. What does the HIPAA Privacy Rule say about a research participant's right of access to research records or results? 2013b. [March 28, 2018]. https://www
.hhs.gov/hipaa /for-professionals /faq/311/what-does-hipaa-say-about-research-participants-right-of-access /index.html. - OHRP (Office for Human Research Protections). Coded private information or specimens use in research, guidance (2008). 2016a. [March 29, 2018]. https://www
.hhs.gov/ohrp /regulations-and-policy /guidance/research-involving-coded-private-information/index.html. - OHRP. Federal policy for the protection of human subjects (“Common Rule”). 2016b. [March 29, 2018]. https://www
.hhs.gov/ohrp /regulations-and-policy /regulations/common-rule/index .html. - OHRP. History: ORRP history. 2016c. [May 29, 2018]. https://www
.hhs.gov/ohrp /about-ohrp/history/index.html. - Pike ER, Rothenberg KH, Berkman BE. Finding fault? Exploring legal duties to return incidental findings in genomic research. Georgetown Law Journal. 2014;102:795–843. [PMC free article: PMC4207216] [PubMed: 25346543]
- Prince AER, Berkman BE. When does an illness begin: Genetic discrimination and disease manifestation. The Journal of Law, Medicine & Ethics. 2012;40(3):655–664. [PMC free article: PMC4142506] [PubMed: 23061591]
- Prucka SK, Arnold LJ, Brandt JE, Gilardi S, Harty LC, Hong F, Malia J, Pulford DJ. An update to returning genetic research results to individuals: Perspectives of the industry pharmacogenetics working group. Bioethics. 2015;29(2):82–90. [PMC free article: PMC4305195] [PubMed: 24471556]
- Ramoni RB, McGuire AL, Robinson JO, Morley DS, Plon SE, Joffe S. Experiences and attitudes of genome investigators regarding return of individual genetic test results. Genetics in Medicine. 2013;15(11):882–887. [PMC free article: PMC4143384] [PubMed: 23639901]
- Ray T. 360Dx. Feb 16, 2018. South Carolina Supreme Court mulls whether Quest is healthcare provider in wrongful death suit.
- Rothstein MA. Ethical issues in big data health research: Currents in contemporary bioethics. The Journal of Law, Medicine & Ethics. 2015;43(2):425–429. [PubMed: 26242964]
- SACHRP (Secretary's Advisory Committee on Human Research Protections). Attachment C: Return of individual results and special considerations of issues arising from amendments of HIPAA and CLIA. 2015. [May 28, 2018]. https://www
.hhs.gov/ohrp /sachrp-committee /recommendations/2015-september-28-attachment-c/index.html. - SACHRP. Attachment B: Return of individual research results. 2016. [July 13, 2017]. https://www
.hhs.gov/ohrp /sachrp-committee /recommendations/attachment-b-return-individual-research-results/index.html. - Wolf SM. The role of law in the debate over return of research results and incidental findings: The challenge of developing law for translational science. Minnesota Journal of Law, Science & Technology. 2012;13(2) [PMC free article: PMC3874275] [PubMed: 24379751] [CrossRef]
Footnotes
- 1
This section draws on a paper commissioned by the Committee on the Return of Individual-Specific Research Results Generated in Research Laboratories, “Analysis of Legal and Regulatory Landscape Relevant to Return of Individual Results Generated from Biospecimens in Research” by Christi J. Guerrini (see Appendix C).
- 2
Changes in the European Union under the new General Data Protection Regulation, which went into effect May 25, 2018, confer a right to access personal data that is being processed, including research data, except in cases where providing access would impair the research (Regulation [EU] 2016/679, article 15).
- 3
This sentence was changed from the prepublication report.
- 4
Alaska Stat. § 18.13.010(a)(2).
- 5
Colo. Rev. Stat. Ann. § 10-3-1104.7(1)(a).
- 6
PHI is defined as individually identifiable health information, which is any information (including genetic information) that (a) is created or received by a covered entity or employer; (b) “relates to the past, present, or future physical or mental health or condition of an individual; the provision of health care to an individual; or the past, present, or future payment for the provision of health care to an individual”; and (c) identifies or could be used to identify the individual (45 C.F.R. § 160.103).
- 7
A designated record set is defined as a group of records maintained by or for a covered entity that comprises the (1) medical records and billing records about individuals maintained by or for a covered health care provider; (2) enrollment, payment, claims adjudication, and case or medical management record systems maintained by or for a health plan; or (3) other records that are used, in whole or in part, by or for the covered entity to make decisions about individuals (45 C.F.R. § 164.501). This includes records used to make decisions about any individuals, whether or not the records have been used to make a decision about the particular individual requesting access.
- 8
HIPAA-covered entities include health plans, health care clearinghouses, and health care providers that transmit health information in electronic form in connection with a covered financial or administrative transactions (e.g., billing transactions). Research laboratories are HIPAA-covered entities if they electronically conduct a covered transaction or if they function as part of a larger covered entity (e.g., hospitals, medical centers). HIPAA also extends to business associates of covered entities (45 C.F.R. § 160.103).
- 9
The final rule to the CLIA Program and HIPAA Privacy Rule amended 42 C.F.R. § 493; 45 C.F.R. § 164.
- 10
42 C.F.R. § 493.2.
- 11
42 C.F.R. § 493.2.
- 12
New York and Washington both have implemented laboratory licensure programs with requirements at least as stringent as those required by CLIA and laboratories in these states are recognized as CLIA exempt. Both states have rules prohibiting the return of research results from non-certified laboratories (CMS, 2017).
- 13
When laboratory testing for clinical trials is not conducted in a CLIA-certified laboratory, other standards, such as good clinical laboratory practices, can be adopted to help to support the quality of those results (Ezzelle et al., 2008).
- 14
42 U.S.C. § 263a(h)–(j); 42 C.F.R. § 493.1806(c)(3)–(d), 493.1834(a)–(d), 493.1846.
- 15
45 C.F.R. § 164.524(a)(1)(iii) (effective to April 7, 2014).
- 16
Although CLIA defines the term “CLIA-exempt” in reference to those laboratories operating in states that have established a CMS-approved licensure program with requirements that are at least as stringent as those under CLIA, for the purposes of the privacy rule's access right, HHS's Office for Civil Rights (OCR) has interpreted the term “CLIA-exempt” to also include research laboratories operating under the CLIA exception (i.e., those that do not require CLIA certification because they conduct tests on human biospecimens for research purposes and do not report patient-specific results for the diagnosis, prevention, or treatment of a disease or the assessment of the health of an individual). This issue is illustrative of the confusion generated by ambiguity in terminology used in regulations and guidance and the need for harmonization of terminology (discussed later in this chapter).
- 17
In 2016, OCR published guidance on the types of information that may be contained within the DRS (HHS, 2016), indicating, for example, that this may include results of genetic testing maintained by or for a clinical laboratory, including gene variant information. However, this guidance did not refer specifically to test results generated by research laboratories. Older guidance that does address obligations of research laboratories pre-dates the 2014 regulatory changes. This earlier guidance states that research participants shall have access to “any research records or results that are actually maintained by the covered entity as part of a designated record set” (OCR, 2013b).
- 18
Testimony of Wendy Chung of Columbia University at the public meeting of the Committee on the Return of Individual-Specific Research Results Generated in Research Laboratories on September 6, 2017.
- 19
45 C.F.R. 46.
- 20
21 U.S.C. § 321(h).
- 21
21 C.F.R. § 812,3(g)(h).
- 22
21 C.F.R. 812.3.
- 23
Testimony of Adam Berger of FDA at an open session of the Committee on the Return of Individual-Specific Research Results Generated in Research Laboratories on July 19, 2017.
- 24
The joint amendment to the CLIA Program and HIPAA Privacy Rule amended 42 C.F.R. § 493.1291; 45 C.F.R. § 164.524.
- 25
45 C.F.R. § 164.514(b).
- 26
45 C.F.R. § 46.101(b)(4) and 45 C.F.R. § 46.102(f) (defining research to involve identifiable private information and describing exemption for research involving existing non-identified data and biospecimens).
- 27
See Appendix C, p. 335, Americans with Disabilities Act.
- 28
See Appendix C, p. 335, Americans with Disabilities Act.
- 29
See Appendix C, p. 336, Americans with Disabilities Act.
- 30
Genetic Information Nondiscrimination Act of 2008, Public Law 110-233, 110th Cong. (May 21, 2008).
- 31
42 U.S.C. § 300gg-4(d).
- 32
The Americans with Disabilities Act (ADA) also prohibits discrimination in employment and other contexts but was enacted to protect individuals with disabilities. It is not clear whether an asymptomatic individual with a genetic predisposition to a condition that has not yet manifested would be considered to have a disability under the ADA.
- 33
However, it should be noted that the Patient Protection and Affordable Care Act (ACA) seemingly contradicts GINA with respect to the release of genetic information by encouraging employers to offer wellness programs, including diagnostic testing programs, that tie health insurance costs to employee program participation (Public Law 111-148 § 1201).
- 34
42 U.S.C. § 2000ff-1(a)–(b).
- 35
42 U.S.C. § 2000ff-5(a)–(b).
- 36
Public Law 111-148 § 1201 (2010) (codified at 42 U.S.C. § 300gg-3).
- 37
A tort is a civil wrong (other than breach of contract) for which a remedy may be obtained, usually in the form of damages (Legal Information Institute, 2018).
- 38
There is, however, an open question of whether a laboratory is itself a health care provider with fiduciary obligations. This question is presented in a pending federal lawsuit in South Carolina, Williams v. Quest Diagnostics, which is based on allegations that a clinical laboratory returned erroneous genetic test results resulting in the death of an individual who received the wrong treatment (Ray, 2018).
- 39
Of note, analysis of and commentary on changes to the interpretation of CLIA regulations were identified by the sponsors as outside the study scope, so such options are not discussed in this chapter.
- 40
As discussed in Chapter 3, external accountability requires a system for independent verification (i.e., inspection by external experts without conflict of interest or intractable bias toward any one investigator or perhaps even bias toward the institution) to determine whether laboratories are adhering to quality standards.
- 41
A third possible approach that has been suggested by some legal scholars is for the federal agencies to exercise enforcement discretion (i.e., not take action against laboratories or institutions that fail to comply with their regulations until the apparent conflict has been resolved) (Barnes et al., 2015). While the committee understands the practicality of this approach in the interim, it believes that the agencies should work to ensure that their regulations do not conflict and not rely on enforcement discretion to resolve apparent conflicts. Additionally, the likely effect of this approach would be that institutions would continue to interpret the regulations in their own way, resulting in inequitable access for research participants.
- 42
45 C.F.R. § 164.103, 45 C.F.R. § 164.105(a)(2)(iii).
- 43
Some research, including some clinical trials, can involve covered transactions such as billing and therefore could not be conducted by the non-covered portion of a hybrid entity.
- 44
Except as permitted by HIPAA—for example, with a valid patient authorization.
- 45
Research results that are also used in clinical decision making (e.g., results from a clinical trial conducted in a health care setting to evaluate the comparative effectiveness of medical treatments) would not be excluded from the designated record set and could be accessed by participants upon request under HIPAA.
- Reshaping the Legal and Regulatory Landscape to Support Return of Individual Res...Reshaping the Legal and Regulatory Landscape to Support Return of Individual Research Results - Returning Individual Research Results to Participants
Your browsing activity is empty.
Activity recording is turned off.
See more...