

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.
This publication is provided for historical reference only and the information may be out of date.

Scope
As the number of large scale virus genome sequencing projects has grown, so too has the need for specialized resources designed to enhance the accessibility and utility of large sequence datasets. Virus Variation is a comprehensive resource designed to support search, retrieval, and display of large virus sequence data sets—providing users with the functionalities necessary to facilitate discovery activities.
This resource includes a search interface through which users can search and retrieve sequences based on a number of biological and clinical criteria. The selected sequences can then be downloaded or analyzed using a suite of Web-based tools and displays.
Currently, three viruses are included within Virus Variation—Dengue, West Nile, and Influenza—with more than 260,000 individual sequences between them. The resource is expanding and new viruses will be added in response to sequencing efforts and public health demand.
History
The Virus Variation Resource is an outgrowth of the NCBI Influenza Virus Resource originally created in 2004 to support the thousands of Influenza virus genomes sequenced during the National Institute of Allergy and Infectious Diseases (NIAID)-initiated Influenza Genome Sequencing Project (1). The goal of the resource then as now was to provide a suite of interfaces and tools designed specifically for large sequence datasets.
The first iteration of the Virus Variation resource was developed around Flaviviruses, with Dengue Virus added in 2009, and West Nile virus two years later (2). The current implementation combines the previous resources into a single comprehensive construct—building upon historic functionalities but flexible enough to accommodate a broad range of viruses.
Data Model
The Virus Variation Resource is comprised of three components: a specialized database, a unique search interface, and a group of sequence displays. The database is loaded with data processed from GenBank records, and virus-specific annotation pipelines are used to produce standardized, consistent protein and gene annotation across all sequences from a given species. Automated and manual procedures capture descriptors—metadata—from sequence records, literature, and other databases, then map these to a common vocabulary, and store them with the sequences they describe.
Stored, standardized sequence data and related metadata provide infrastructure for an enhanced search interface that allows users to retrieve and download protein and nucleotide sequence sets based on a variety of biological criteria—like protein or gene of interest, genotype, host, collection country or region, disease severity, and collection date—as well as sequence patterns and key word searches. Specialized tools including a multi-sequence alignment viewer and phylogenetic tree builder use precalculated alignments to rapidly analyze sequences selected by the user and retrieved from the database.
Dataflow
Sequence Annotation Pipeline
Annotation vagaries and inconsistencies are a major impediment to sequence analysis. Virus Variation mitigates this problem using standardized sequence annotation pipelines that provide consistent annotation across all sequences belonging to a given viral species. Reference sequence sets are used to annotate proteins and other biologically and clinically relevant features. For example, the flu annotation pipeline generates information about drug-resistance mutations and the completeness of nucleotide and coding region sequences; both are stored in the database.
In general, the pipelines for each virus loaded into Virus Variation use a common backbone but unique reference protein sets and parsing strategies. For example, in the Dengue annotation pipeline the incoming sequence is initially assigned a genotype using megaBlast and a reference sequence set. That genotype assignment then points the annotation pipeline to a specific set of reference proteins that are used to annotate the new sequence. The annotation pipelines are used for both internal database loading and as a public resource for Influenza virus—providing standardized annotation for some GenBank submissions.
GenBank Submission Pipeline for Influenza Viruses
NCBI is a collaborator with the NIAID Influenza Genome Sequencing Project and has been tasked with gathering Influenza sequences and related metadata from J. Craig Venter Institute (JCVI), annotating the sequences, and releasing them in GenBank. NCBI has created an automated pipeline to facilitate the large number of sequences generated from the project.
In the pipeline, metadata are retrieved and updated daily from a JCVI ftp site and loaded onto an internal NCBI database. NCBI works closely with JCVI, viral sample providers, and the influenza virus research community in establishing the minimum and optional metadata sets to be incorporated into GenBank records. Organism names for new virus isolates are entered in the NCBI Taxonomy Database. NCBI staff also manually review the metadata and communicate with data providers if there are any issues.
Sequencing data are assembled at JCVI and consensus sequences are verified with the Influenza Virus Genome Annotation Tool (FLAN, for FLu Annotation, see the Virus Genome Processing and Tools Chapter) and then submitted to NCBI through ftp once they are error free. At NCBI, the sequences are processed by FLAN and feature tables generated. These are combined with associated metadata to create GenBank files. In the past 8 years, nearly 11,200 complete influenza virus genomes have been generated from the NIAID project, and published in GenBank.
Database Loading Pipeline
The database loading pipeline is an automated process that parses data from records available in GenBank and maps them to fields used in the Virus Variation database. This process uses generalized parsing strategies to capture both common biological data like host and country of origin, as well as individualized strategies to capture more specific—often clinically relevant—data associated with particular viruses.
The loading pipelines are dependent on vocabulary lists that allow mapping of data parsed from records to controlled descriptors used within the database and displays. For example, host names—including common names and misspelled names—are mapped with these vocabulary lists to scientific names associated with taxonomy IDs in the NCBI Taxonomy Database and host group names like “birds” or “mammals” used in the search pages.
These automated processes are augmented by manual operations based on literature and semi-automated procedures used to capture third-party data releases. Annotation and data capture is also facilitated by community outreach efforts that seek to develop standard, experimentally-driven gene models and reference protein sets. These efforts also encourage the inclusion of rich metadata sets in public database submissions, as well as metadata sharing.
Database
The Virus Variation database stores sequence information derived from the annotation pipeline and associated metadata describing the sample in standardized formats. To balance storage flexibility with efficient data retrieval, Virus Variation combines a relational database with documents containing raw data.
Curation Interface
Since the Virus Variation database loading procedure uses a hybrid of automated and manual procedures, it is important that NCBI staff have the ability to review sequences with loading errors as well as enter data manually into the database. The Virus Variation curation interface enables curators to filter and sort sequences based on virus type, loading errors, and on a number of descriptors like sequence length. A number of editable fields are displayed for each sequence—such as country, isolation date, and host—in a generalized format that is the same for each virus. Curators can review data associated with a given sequence and enter data manually into these fields as guided by literature or other sources. Additionally, there is the ability to adjust the displayed fields and messages to fit the needs of specific viruses and/or database loading procedures.
Access
The Virus Variation Resource can be accessed at http://www.ncbi.nlm.nih.gov/genomes/VirusVariation/. This home page includes links to virus-specific modules.
Search Interface
The unique search interface allows users to construct database queries based on a number of criteria including gene or protein region, GenBank accessions, and keywords, as well as biologically relevant descriptors like disease associations, host organism, and geographic information about the sample. Although the same basic interface design is used throughout the resource, the interface is customized to include specific search fields for individual viruses. The number of sequence records retrieved by a search is displayed with the query builder frame of the page—so that the user can modify search parameters. Once the desired query is built, retrieved sequences can be downloaded in a variety of formats directly or can be displayed within the results page.
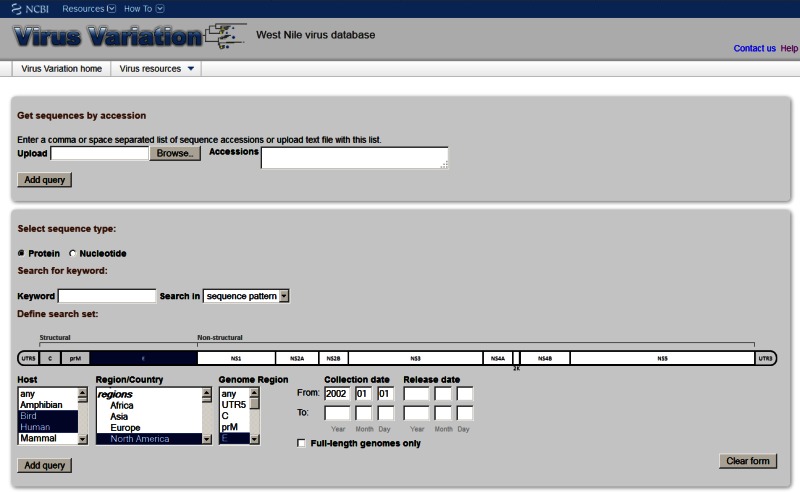
Figure 1.
The Virus Variation search interface. Users can use a number of search criteria including sequence patterns, host, geographic region, and collection date to retrieve either protein or DNA sequences from specified genome regions.
Results Page
The results page displays all the sequences retrieved in a given search where individual sequences can be selected prior to subsequent analysis or download. Individual records can be sorted by a variety of descriptors, selected or deselected, downloaded, sent to the multi-sequence alignment viewer, or sent to the phylogenetic tree viewer.
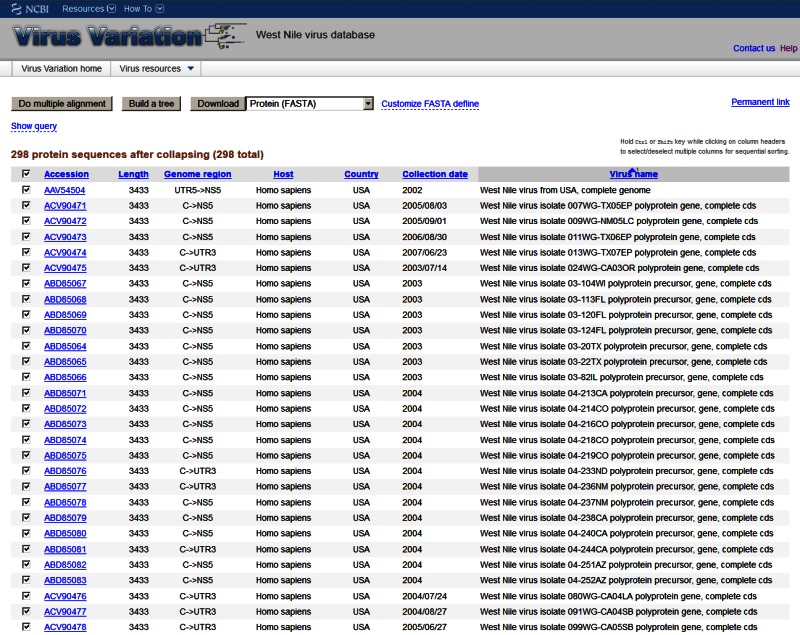
Figure 2.
The Virus Variation search results page. Records retrieved during a search can be displayed within the results page where individual sequences can be selected for download or further analysis.
Multi-sequence Alignment Viewer
The multi-sequence alignment viewer allows users to display alignments of selected protein or nucleotide sequences. Alignments are precalculated to save processing time. The viewer is based on the Genome Workbench alignment viewer and includes a number of advanced features including multiple display and scoring options. In the default view a consensus sequence is displayed as the anchor, and a reference feature table is used to define protein (or gene) positions and other important landmarks. The displayed features facilitate navigation along the alignment and allow users to hone in on regions of interest. The anchor sequence can be changed from a consensus sequence to any in the alignment to facilitate greater scrutiny of specific sequences within the alignment.

Figure 3.
The Virus Variation multiple sequence alignment viewer. Selected protein or nucleotide sequences can be displayed in precalculated alignments allowing rapid comparison of sequences.
Phylogenetic Tree Viewer
The phylogenetic tree viewer displays phylogenetic trees built from alignments of sequences selected in the results page. The current viewer includes collapsible leaves, which allows a user to adjust the resolution of a selected subtree to improve viewing of large data sets (3). Users can also markup sequences based on date range and search for and tag sequences based on country, host, accession, and other descriptors. This feature provides a graphic representation of the metadata associated with sequences, enhancing the user’s ability to make associations between phylogenetics and sequence descriptors.

Figure 4.
The Virus Variation phylogenetic tree viewer. Selected nucleotide and protein sequences can be quickly displayed on phylogentic trees using precalculated alignments and a variety of clustering and distance algorithms. Sequences can be searched by metadata, such as country, host, and accession, and marked up in green. Sequences from specific dates can also be highlighted in red.
References
- 1.
- Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. The influenza virus resource at the National Center for Biotechnology Information. J Virol. 2008 Jan 8;2(2):596–601. [PMC free article: PMC2224563] [PubMed: 17942553]
- 2.
- Resch W, Zaslavsky L, Kiryutin B, Rozanov M, Bao Y, Tatusova TA. Virus variation resources at the National Center for Biotechnology Information: dengue virus. BMC Microbiol. 2009 Apr 2;9:65. [PMC free article: PMC2675532] [PubMed: 19341451]
- 3.
- Zaslavsky L, Bao Y, Tatusova TA. Visualization of large influenza virus sequence datasets using adaptively aggregated trees with sampling-based subscale representation. BMC Bioinformatics. 2008 May 16;9:237. [PMC free article: PMC2416652] [PubMed: 18485197]
- Virus Variation - The NCBI HandbookVirus Variation - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...