I-type lectins are defined as glycan-binding proteins (excluding antibodies and T-cell receptors) in which the binding domain is homologous to the large and varied immunoglobulin superfamily (IgSF) of proteins. Among I-type lectins, the Siglec family of sialic acid–recognizing lectins is the best characterized subgroup, both structurally and functionally, and is therefore the major focus of this chapter. Details of their discovery, characterization, binding properties, and biology are provided, along with discussions of their functional roles in vertebrate biology, with most currently available information being in mammals, and multiple unusual changes during human evolution.
HISTORICAL BACKGROUND AND OVERVIEW
The Ig fold is made up of antiparallel β-strands organized into a β-sandwich containing 100–120 amino acids and usually stabilized by an intersheet disulfide bond. Three types or “sets” of Ig domains are defined based on homologies in sequence and structure to domains of antibodies: the V-set variable-like domain, the C1- and C2-set constant-like domains, and the I-set domain that combines features of both V- and C-set domains.
Before the 1990s, it was thought that antibodies were the only IgSF members capable of recognizing glycans. The first direct evidence for nonantibody IgSF glycan-binding proteins came from independent studies on sialoadhesin (Sn), a sialic acid (Sia)–dependent binding receptor on mouse macrophage subsets, and on CD22, a molecule previously cloned as a B-cell marker. Various techniques showed that Sn functions as a lectin, including loss of binding following sialidase treatment of ligands, inhibition assays with sialylated compounds, and Sia-dependent binding of the purified receptor to glycoproteins and to red blood cells derivatized to carry Sias in different linkages. With recombinant CD22, abrogation of cell adhesive interactions by sialidase treatment led to the discovery that it was a Sia-binding lectin, with a high degree of specificity for α2-6-linked Sias. Cloning of Sn then showed that it was an IgSF member sharing homology with CD22 and with two other previously cloned proteins, CD33 and myelin-associated glycoprotein (MAG). Demonstration of Sia recognition by CD33 and MAG resulted in the definition of a new family of Sia-binding molecules, which were initially called “sialoadhesins.” Meanwhile, preliminary evidence for glycan binding by additional IgSF members emerged, and a suggestion was made to classify all these molecules as “I-type” lectins. However, it became clear that these four Sia-binding molecules were a distinct subgroup sharing both sequence homology and Ig-domain organization, and that they were not all involved in adhesion. The term Siglec (sialic acid–binding immunoglobulin-like lectin) was therefore proposed in 1998. Subsequently, most of the CD33-related Siglecs (CD33rSiglecs) were discovered as a result of genomic and transcriptomic sequencing projects, which allowed in silico identification of novel Siglec-related genes and cDNAs.
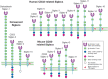
Siglecs are divided into two major subgroups based on sequence similarity (Figure ) and on conservation among mammalian species. The first group comprises Sn (Siglec-1), CD22 (Siglec-2), MAG (Siglec-4), and Siglec-15, for which there are clear-cut orthologs in all mammalian species examined and which share only ∼25%–30% sequence identity among each other. The second group comprises the CD33rSiglecs, which share ∼50%–80% sequence similarity but appear to be evolving rapidly and undergoing exon shuffling and gene conversions of Ig-domain-encoding exons, making it difficult to define orthologs, even between rodents and primates (see below). For this reason, Siglec nomenclature uses numbers and letters to differentiate between some nonhomologous human and mouse Siglecs. In retrospect, it turned out that many distantly related mammals have more clear-cut orthologs, and it is the rodent lineage that “discarded” these genes.
Domain structures of the known Siglecs in humans and mice. There are two subgroups of Siglecs: One group contains sialoadhesin (Siglec-1), CD22 (Siglec-2), MAG (Siglec-4), and Siglec-15, and the other group contains CD33-related Siglecs. In humans, Siglec-12 (more...)
I-TYPE LECTINS OTHER THAN SIGLECS
Several IgSF members other than Siglecs have been proposed to bind glycans or glycoconjugates. The best evidence is for paired immunoglobulin-like type 2 receptors (PILR-α and PILR-β), both of which have a single V-set domain similar to that of Siglecs with a structurally defined Sia-binding site as well as a protein interaction site. PILR proteins bind a subset of mucin-like O-glycosylated membrane proteins involving simultaneous recognition of both the peptide backbone and Sia to mediate high-affinity interactions. Evidence suggests a role of PILRs in the control of innate immunity and host defense against microbial pathogens. PILRs are expressed on immune cells of the myeloid lineage, and PILR-β is also found on NK cells. PILR-α contains two immunoreceptor tyrosine-based inhibitory motifs (ITIMs) and delivers inhibitory signals by recruitment of inhibitory phosphatases. In contrast, PILR-β associates with DNAX activation protein-12 (DAP12), an adapter protein that has an immunoreceptor tyrosine-based activating motif (ITAM) and promotes innate immune cell activation. Platelet endothelial cell adhesion molecule (PECAM)-1 is composed of six extracellular Ig-like domains and selectively recognizes α2-6-linked Sia. PECAM-1 is broadly expressed on endothelial cells and leukocytes and plays multiple roles linked to leukocyte transendothelial migration, inflammation, and vascular biology. The neural cell adhesion molecule (NCAM) and basigin (CD147) have been claimed to recognize and bind oligomannose-type N-glycans on adjacent glycoproteins in the nervous system. NCAM contains five Ig-like domains followed by two fibronectin type III-like domains, and exerts essential functions in the development, plasticity, and regeneration of the nervous system. The different isoforms of basigin contain variable numbers of Ig-like domains and are widely expressed in different tissues where they have roles in reproduction, nervous system function, and immunity. Intercellular adhesion molecule (ICAM)-1 comprises five Ig-like domains and binds numerous ligands, including the integrins LFA-1 and Mac-1, hyaluronan and possibly certain mucin-type glycoproteins. Its expression on leukocytes and endothelial cells is enhanced on cellular activation. ICAM-1 plays crucial roles in cell–cell interactions, extravasation, inflammation, and host defense. Hemolin is an IgSF plasma protein from lepidopteran insects that binds lipopolysaccharide (LPS) from Gram-negative bacteria and lipoteichoic acid from Gram-positive bacteria. Hemolin appears to have two binding sites for LPS—one that interacts with the phosphate groups of lipid A and another that interacts with the O-specific glycan antigen and the outer-core glycans of LPS. There is indirect and less convincing evidence for interactions of other IgSF molecules with glycans, such as the peripheral myelin protein P0 with HNK-1, CD83 with Sias, and CD2 with Lewis x (Lex). Overall, with the exception of the Siglecs and the PILRs, the direct assignment of an IgSF fold as the actual binding pocket for glycans has not been achieved. The rest of this chapter is devoted to Siglecs, the best-characterized I-type lectins.
COMMON FEATURES OF SIGLECS
The Amino-Terminal V-Set Sialic Acid–Binding Domain
All Siglecs are type-1 membrane proteins with a Sia-binding, amino-terminal V-set domain and varying numbers of C-set Ig domains that act as spacers, projecting the Sia-binding site away from the plasma membrane. The V-set domain and the adjacent C-set domain contain a small number of invariant amino acid residues, including an “essential” arginine on the F β-strand required for Sia binding, and an unusual organization of cysteine residues. Instead of the typical intersheet disulfide bond between the B and F β-strands, the V-set domain of Siglecs displays an intrasheet disulfide bond between the B and E β-strands, permitting increased separation between the β-sheets. The resulting exposure of hydrophobic residues allows specific interactions with constituents of Sia ligands. All Siglecs studied so far also appear to contain an additional unusual disulfide bond between the V-set domain and the adjacent C-set domain, which would be expected to promote tight packing at the interface between the first two Ig domains. Although the significance of this bond for ligand recognition is unclear, optimal Sia-binding activity of many Siglecs requires the adjacent C-set domain, probably for correct folding and stability.
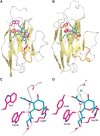
Three-dimensional structures for the mouse Sn, human Siglec-7, and human CD33 V-set domains, the V-set and adjacent C-set domain of Siglec-5, first three Ig-like domains of human CD22, and all five Ig-like domains of mouse MAG have been determined by X-ray crystallography, in the presence and absence of Sia ligands (Figure ). Along with the nuclear magnetic resonance (NMR) structure of Siglec-8, structural templates for Sia recognition by Siglecs appear to be shared among Siglecs. In all instances, an “essential” arginine residue is located in the middle of the F β-strand, making a bidentate salt bridge with the carboxylate of Sia. Siglecs also contain conserved hydrophobic amino acids on the A and G β-strands that interact with the N-acetyl group and glycerol side chain of Sia, respectively. Although all Siglecs appear to use a common template for recognizing Sias, their binding preferences for extended glycan chains vary greatly. The peptide loop between the C and C′ β-strands is highly variable among Siglecs and has a key role in determining their fine sugar specificity. For example, molecular grafting of the C–C′ loop between Siglec-7 and Siglec-9 resulted in switched sugar-binding specificities. Structural studies have shown that this loop appears to be highly flexible, being able to make specific and varied interactions with long glycan chains. In addition, V-set domain sequences are the most rapidly evolving regions in these molecules, likely explaining species differences in binding specificity.
Structural basis of Siglec binding to ligands. X-ray crystal structures of the V-set domains of sialoadhesin (Sn) (A) and Siglec-7 (B) are shown complexed with sialic acid. (C,D) Molecular details of the interactions of sialic acid with Sn and Siglec-7. (more...)
Masking and Unmasking
The cell-surface glycocalyx of most vertebrate cells is richly decorated in glycoconjugates that contain Sias (Chapter 15). The high local concentration of Sias is likely to greatly exceed the Kd value of each Siglec, resulting in self-binding to sialoglycans on the same membrane (cis) that can be directly functional or that can “mask” the Sia-binding site from interacting with sialoglycans on other cells (trans). Consequently, the Sia-dependent binding activity of most naturally expressed Siglecs is strongly enhanced following treatment of cells with sialidase to eliminate the cis-interacting sialylated glycans. A notable exception is Sn, which was discovered via its native property as a Sia-dependent cell-adhesion molecule on macrophages. The “masked” state of most Siglecs is a dynamic equilibrium with multiple cis ligands. Thus, an external probe, a cell surface or a pathogen bearing high-affinity ligands or very high densities of Sia residues, can effectively compete for binding to “masked” Siglecs. In addition, changes in expression of glycosyltransferases or sialidases of endogenous or exogenous origin could influence masking and unmasking of Siglecs at the cell surface, especially during immune and inflammatory responses.
Cell Type–Restricted Expression
Siglecs show restricted patterns of expression in single or related cell types (see below). This is most striking for the conserved Siglecs—Sn (macrophage), CD22 (B lymphocyte), MAG (myelin-forming cell), and Siglec-15 (osteoclast). This theme extends to some of the CD33rSiglecs, most notably in humans, including Siglec-6 on placental trophoblasts, Siglec-7 on NK (natural killer) cells, and Siglec-11 and -16 on tissue macrophages, including brain microglia in humans. In the mouse, Siglec-H and CD33 are excellent markers of plasmacytoid dendritic cells (DCs) and blood neutrophils, respectively, and Siglec-F is a useful marker of eosinophils and alveolar macrophages. These cell type–restricted expression patterns are thought to reflect discrete, cell-specific functions mediated by each of these Siglecs. However, certain key cells of the human immune system, such as monocytes and conventional DCs, express multiple CD33rSiglecs.
Tyrosine-Based Signaling Motifs
Most Siglecs have one or more tyrosine-based signaling motifs in their cytoplasmic tails or associate with membrane adaptor proteins containing cytosolic tyrosine motifs. The most prevalent motif is the ITIM with the consensus sequence (V/I/L)XYXX(L/V), where X is any amino acid. Up to 300 ITIM-containing membrane proteins are identified in the human genome, and many of these are established inhibitory receptors of the hematopoietic and immune systems. Following tyrosine phosphorylation by Src family kinases, they function by recruiting and activating SH2-domain-containing effectors, especially the protein tyrosine phosphatases SHP-1 and SHP-2. These counteract activating signals triggered by receptors containing ITAMs. Some Siglecs, notably Siglecs-14, -15, -16, and -H and mouse CD33, have a positively charged residue within the transmembrane region, which can associate with the DAP12 ITAM-containing adaptor, thereby mediating activating functions.
EXPRESSION PATTERNS AND FUNCTIONS OF THE CONSERVED SIGLECS
Sialoadhesin (Sn, Siglec-1, CD169)
Sn (CD169) was identified as a Sia-dependent sheep erythrocyte receptor expressed by mouse stromal macrophages. Sn has an unusually large number of Ig domains (seventeen), which are conserved among mammals and reptiles. These may be important for extending the Sia-binding site away from the plasma membrane to promote intercellular interactions. Sn prefers α2-3-linked Sias more than α2-6- and α2-8-linked Sias, and does not bind Sias modified by hydroxylation (Neu5Gc) or side-chain O-acetylation (e.g., Neu5,9Ac2). Notably, this preference is very similar to the pattern of Sias expressed on commensal/pathogenic microbes (Neu5Ac>>Neu5Gc and α2-3>α2-8>>>α2-6).
In humans and mice, Sn expression appears specific for macrophage subsets, especially those resident in lymphoid tissues. These cells play important roles in antigen presentation to B cells and NKT cells, tolerance of self-reactive T cells but also as Trojan horses for viral infections to permit protective immune responses. Sn can also be strongly induced on monocytes, macrophages, and monocyte-derived DCs in vitro by type I interferons or agents such as viruses and Toll-like receptor (TLR) ligands that induce interferon production. Accordingly, Sn is up-regulated on circulating monocytes in human immunodeficiency virus (HIV)-infected individuals and on macrophages in patients with rheumatoid arthritis, primary biliary cirrhosis, and systemic lupus erythematosus (SLE). Sn expression on inflammatory macrophages has been associated with a favorable prognosis in colorectal cancer and with more severe disease in proliferative glomerulonephritis. Many of the above disease associations may reflect exposure of macrophages to interferons rather than being causally related. Indeed, in the BWF1 murine model of spontaneous SLE, there was no influence of Sn deficiency on disease severity. However, in mouse models of inherited neuropathy, autoimmune uveoretinitis, and experimental allergic encephalomyelitis (EAE), Sn-deficient mice showed reduced inflammation accompanied by reduced levels of T-cell and macrophage activation. These results suggest that Sn may suppress regulatory T-cell (Treg) expansion thereby promoting inflammation. Sn can also efficiently mediate the capture and uptake of exosomes released from B lymphocytes following apoptosis and therefore play a role in antigen presentation.
A role for Sn in phagocytic interactions of macrophages with various sialylated bacteria and protozoal pathogens has also been shown, including Neisseria meningitidis, Campylobacter jejuni, and Trypanosoma cruzi. A role in host protection was observed using an infection model with Group B streptococcus (GBS), in which Sn-deficient mice showed increased bacterial spread. Conversely, Sn expression on macrophages and monocyte-derived DCs can be exploited by enveloped viruses displaying host-derived Sias, leading to their capture, uptake, and dissemination. This mechanism was first seen with the porcine reproductive and respiratory syndrome virus, which targets lung alveolar macrophages of pigs, and more recently with HIV and other retroviruses. On HIV, Sn can recognize both sialylated gp120 glycoprotein and GM3, a monosialylated ganglioside terminating in Neu5Acα2-3Gal. GM3 is packaged into HIV during the budding from infected cells which occurs in lipid rafts. On monocyte-derived DCs, Sn interactions with GM3 on HIV lead to membrane invaginations containing viral particles that are very efficiently transferred to T cells in a process known as “trans infection.”
CD22 (Siglec-2)
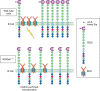
CD22 is a developmentally regulated cell-surface glycoprotein on B cells, expressed at approximately the time of Ig gene rearrangement and lost when mature B cells differentiate into plasma cells. CD22 has seven Ig-like domains and the intracellular region has six tyrosine-based signaling motifs, three of which could function as ITIMs. CD22 is a well-established negative regulator of B-cell activation, making an important contribution toward the threshold for signaling via the B-cell receptor (BCR) complex. Following BCR cross-linking, CD22 is rapidly tyrosine-phosphorylated on its ITIMs by the protein tyrosine kinase Lyn. This leads to recruitment and activation of the SHP-1 tyrosine phosphatase and subsequent inhibition of downstream signaling mediated via the BCR. Although some activating molecules are also recruited to the phosphorylated tyrosine motifs in CD22, the net phenotype of CD22-deficient mice is consistent with a primary role of CD22 in negative regulatory signaling, manifested by enhanced BCR-induced calcium signaling, enhanced B-cell turnover, reduced numbers of recirculating B cells in the bone marrow, and reduced numbers of marginal zone B cells.
Of all the Siglecs, CD22 has the most conserved specificity for sialylated ligands, binding primarily to α2-6-linked Sias of the type Neu5Ac(or Neu5Gc)α2-6Galβ1-4GlcNAc, which are common capping structures of many complex-type N-glycans. Additional specificity can be conferred by the nature of the Sia moiety as well as sulfation of the underlying glycan. Neither human nor mouse CD22 binds 9-O-acetylated Sias; mouse CD22 has a strong preference for Neu5Gc over Neu5Ac, whereas human CD22 binds both of the latter forms. Recombinant soluble CD22 can precipitate a subset of glycoproteins from B-cell lysates including CD45, a major sialoprotein of T and B cells. However, on B cells, CD22 appears to mainly be either cis-associated with other CD22 molecules in a glycan-dependent manner, or with the BCR, which it inhibits in a glycan-independent manner. These interactions are consistent with studies of mouse mutants that either lack CD22 glycan ligands or express mutated forms of CD22 unable to bind glycans. B cells from ST6Gal-I-deficient mice that lack CD22 ligands show an anergic phenotype, essentially the opposite of the phenotype observed in CD22-deficient mice. Mice expressing a lectin-inactive version of CD22 with a mutated binding site arginine also show reduced BCR signaling (anergy) and, similar to ST6Gal-I-deficient mice, they show increased CD22-BCR association and stronger CD22 phosphorylation (Figure ). Likewise, Cmah-null mice deficient in Neu5Gc have reduced ligands for mouse CD22 and Siglec-G (see below) and show B cell hyperactivity.
Proposed biological functions mediated by CD22. CD22 glycan-dependent homotypic interactions in equilibrium with CD22–BCR interactions. CD22 is clustered together by Sia-dependent homotypic interaction and kept away from BCR on wild-type B cells, (more...)
Besides regulating B-cell functions via cis-interactions, CD22 can also mediate trans-interactions with sialylated ligands on other cells that sequester CD22 away from the BCR. This could be important for raising B-cell activation thresholds to “self”-antigens and may help to ensure that signaling through the BCR can only occur in lymphoid tissues in which CD22 α2-6-sialylated ligands are abundant. Although CD22-deficiency alone does not lead to extensive autoimmune reactions, mice deficient in both CD22 and the other major B-cell Siglec, Siglec-G, develop SLE-like symptoms, including production of IgG autoantibodies and glomerulonephritis.
The restricted expression and properties of CD22 make it an attractive therapeutic target. Antibody-based CD22-targeting agents, such as antibody–drug conjugate inotuzumab ozogamicin and recombinant antibody–toxin fusion protein moxetumomab pasudotox, have been approved for clinical use (for acute B-cell leukemia and hairy cell leukemia, respectively).
Myelin-Associated Glycoprotein (Siglec-4)
MAG, a minor constituent of central nervous system (CNS) and peripheral nervous system (PNS) myelin, has five Ig-like domains and is well conserved among vertebrates. It is expressed by myelin-forming cells: oligodendrocytes in the CNS and Schwann cells in the PNS. In mature myelinated axons it is found primarily on the innermost (periaxonal) myelin wrap, directly across from the axon, but not in the multilayers of compacted myelin. MAG-deficient mice develop normal myelin, but defects in myelin and axons increase as animals age, indicating a role for MAG in the maintenance of myelinated axons, rather than in the process of myelination. MAG-null mice display late-onset axonal degeneration leading to progressive loss of motor function. MAG-null mice fail to show characteristic myelin-induced increases in neurofilament phosphorylation and therefore reduced axon diameters, indicating that MAG signaling is required for optimal myelin–axon interactions. Mendelian disorders (autosomal recessive spastic paraplegia 75) caused by homozygous mutation in the MAG gene, including a point mutation in its Sia-binding arginine, have been reported, and clinical features of the disease are consistent with the roles of MAG revealed by the studies of Mag-deficient mice. MAG also directly inhibits neurite outgrowth from a wide variety of neuronal cell types in vitro. This contributes to the inhibitory activity of myelin on axon outgrowth after nervous system injury, hampering functional recovery.
Genetic and biochemical evidence indicate that gangliosides (sialylated glycolipids) are important physiological ligands for MAG, mediating both myelin–axon stability and inhibition of axon outgrowth. Recombinant forms of MAG bind selectively to the abundant axonal gangliosides GD1a and GT1b (Chapter 11). The phenotype of MAG-deficient mice is similar to that of mice lacking an N-acetylgalactosaminyltransferase (coded by the B4galnt1 gene) required for synthesis of GD1a and GT1b, resulting in progressive motor dysfunction. Notably, human mutations in the same gene (B4GALNT1) result in hereditary spastic paraplegia, a progressive motor neuropathy reminiscent of that of B4galnt1-null mice. Binding of soluble MAG to some types of neurons, and subsequent MAG-mediated inhibition of neurite outgrowth, is Sia and ganglioside dependent. MAG-mediated inhibition of neurite outgrowth from B4galnt1-null mouse neurons is diminished, whereas MAG still inhibits neurite outgrowth from the neurons of mice lacking the “b-series” gangliosides (GD3 synthase-null; Chapter 11), which lack GT1b but express GD1a. These findings suggest that gangliosides GD1a or GT1b act as functional docking sites for MAG on neuronal cells. MAG binds to other axonal receptors including a family of GPI-anchored proteins (Nogo receptors NgR1 and NgR2) and paired immunoglobulin-like receptor B (PirB). Gangliosides, NgRs, and PirB may act independently or interactively as MAG receptors, linking MAG binding to axonal signaling in different neuronal cell types.
Siglec-15
Siglec-15 was first described as a highly conserved ancient Siglec in vertebrates. It lacks the typical arrangement of cysteines seen in the V-set Ig domain of other Siglecs and has an unusual intron–exon arrangement. Nevertheless, it can bind the Sialyl-Tn structure (Neu5Acα2-6GalNAcα) and other structures containing a Neu5Acα2-6HexNAc determinant. It associates with DAP12 but also has an ITIM-like motif in its cytoplasmic tail. Although first reported on macrophages and DCs in human lymphoid tissues, Siglec-15 is most strongly expressed in osteoclasts and their precursors in which it plays an important role, together with receptor activator of nuclear factor (NF)-κB (RANK), in triggering osteoclast differentiation. Osteoclasts are key cells involved in bone degradation and share a common hemopoietic progenitor with macrophages. Mice lacking Siglec-15 show a mild osteopetrosis and impaired osteoclast differentiation. Specific antibodies against Siglec-15 phenocopy this state, because of antibody-induced internalization and degradation of Siglec-15. In addition, Siglec-15 is expressed in some tumor tissues (tumor-associated macrophages and cancer cells), and an antibody against Siglec-15 was shown to suppress the growth of a mouse melanoma in vivo. Siglec-15 therefore provides a novel target for diseases, such as menopause-related osteoporosis and cancer.
GENOMIC ORGANIZATION, EXPRESSION PATTERNS, AND FUNCTIONS OF THE CD33-RELATED SIGLECS
Genes encoding most of the CD33rSiglec subfamily are clustered on human chromosome 19q13.3-13.4 or the syntenic region of mouse chromosome 7. They include CD33 (Siglec-3), Siglecs-5 through -12, Siglec-14, and Siglec-16 in humans and CD33 and Siglecs-E, -F, -G, and -H in mice. Similar clusters are found in other mammals. It is difficult to assign all definitive orthologs between primates and rodents, resulting in different nomenclatures. One reason is that most IgSF domains are encoded by exons with phase-1 splice junctions, allowing exon shuffling without disrupting open reading frames, resulting in hybrid genes that are difficult to distinguish from similarly organized genes in other species. A second reason is that the Sia-binding V-set domains of the CD33rSiglecs are rapidly evolving, presumably to adjust their binding specificity to the rapid evolution of the endogenous host sialome, as well as evasion of binding by pathogens via molecular mimicry (Chapter 15) or specific protein-mediated interactions (see below). There are also multiple gene conversion events between adjacent genes and pseudogenes within this cluster. Of interest is the finding that humans show many CD33rSiglec differences compared with our closest evolutionary cousins (the chimpanzees), more than the differences between mice and rats, which shared a common ancestor much earlier (see below).
As mentioned earlier, an “essential” arginine residue in all known Siglecs is required for binding Sia-containing ligands. This residue is often mutated in nature, resulting in loss of binding ability. Examples include Siglec-12 in humans, Siglecs-5 and -14 in the chimpanzee, gorilla, and orangutan, Siglec-6 in the baboon, and Siglec-H in the rat. The common arginine codon (CGN, where N is any nucleotide) tends to be highly mutable because of the CpG sequence (which is prone to TpG or CpA transition via cytosine methylation–deamination). However, the frequency with which such events occur suggests that it might be a natural mechanism to eliminate Sia binding of a given Siglec, when such activity becomes inappropriate under changing evolutionary pressures, without causing a complete loss of the Siglec. Overall, it appears that this class of SIGLEC genes is subject to multiple “Red Queen” effects in evolution, in which evolutionary changes of sialyltransferases in response to the emergence of Sia-binding pathogens may lead to subsequent evolutionary changes of Siglec specificities (Figure ).
Probable evolutionary chain of Red Queen effects involving Sias and CD33rSiglecs. See text for discussion. (Redrawn, with permission, from Padler-Karavani V, et al. 2014. FASEB J
28: 1280–1293.) To limit further complexity, two additional Red (more...)
Below, we provide a brief summary of the main features of the human CD33rSiglecs and, where relevant, their murine counterparts.
CD33 (Siglec-3)
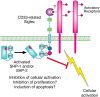
CD33 is a marker of early human myeloid progenitors and leukemic cells and is also expressed on monocytes and tissue macrophages, including brain microglia. It has two Ig domains and was the first of the CD33rSiglecs to be characterized as an inhibitory receptor, suppressing activation of FcγRI and recruiting SHP-1 and SHP-2 (Figure ). CD33 has some preference for α2-6- rather than α2-3-sialylated glycans and binds strongly to sialylated ligands on myeloid leukemia cell lines. The restricted expression of CD33 has been exploited in the treatment of acute myeloid leukemia using gemtuzumab ozogamicin, a humanized anti-CD33 monoclonal antibody coupled to the toxic antibiotic calicheamicin. Binding of anti-CD33 mAbs to CD33 triggers endocytosis of the bound antibody. This depends on ITIM phosphorylation, recruitment of the E3 ligase Cbl, and ubiquitylation of the CD33 cytoplasmic tail. Selective expression of CD33 on leukemic progenitor cells also makes it an attractive target for therapy using chimeric antigen receptors expressed on cytotoxic T cells.
Proposed biological functions mediated by CD33-related Siglecs. A generic CD33-related Siglec is represented, showing the location of the immunoreceptor tyrosine-based inhibitory motif (ITIM) and the potential for inhibitory signaling.
Recently, two coinherited single-nucleotide polymorphisms (SNPs) have been associated with protection of humans against late-onset Alzheimer's disease. These SNPs result in increased exon 2 skipping, leading to raised levels of CD33 lacking the V-set domain and reduced levels of full-length CD33. Because full-length CD33 can inhibit microglial cell uptake of Aβ protein in a Sia-dependent manner, it is thought that individuals lacking the protective SNPs may accumulate more toxic Aβ proteins, thus driving Alzheimer's disease pathology. Targeting CD33 using antibodies that either inhibit function or promote internalization and degradation may be a useful approach to Alzheimer's disease. A complexity arises because the truncated form of CD33 appears to be retained intracellularly in peroxisomes, and the biological consequence of this diversion is currently uncertain.
The murine ortholog of CD33 exists as two spliced forms that differ in the cytoplasmic region, neither containing the typical ITIM found in most other CD33rSiglecs. Furthermore, mouse CD33 has a lysine residue in the transmembrane sequence and couples to the DAP12 transmembrane adaptor, as shown for mouse Siglec-H and human Siglecs-14, -15, and -16. In contrast to human CD33, mouse CD33 in the blood is expressed mainly on neutrophils rather than monocytes, and at low levels in microglia, which also suggests a nonconserved function of this receptor.
Siglec-5 (CD170) and Siglec-14
The SIGLEC5 and SIGLEC14 genes are adjacent to each other on chromosome 19 and encode proteins containing four and three Ig-like domains, respectively. Because of ongoing gene conversions within most taxa, the first two Ig domains of Siglec-5 and Siglec-14 share >99% sequence identity but then diverge, with Siglec-5 being an inhibitory receptor with typical ITIMs, whereas Siglec-14 can complex with DAP12 and activate signaling. Both Siglec-5 and Siglec-14 bind similar ligands, with some preference for the sialyl-Tn structure (Neu5Acα2-6GalNAcα) and α2-8-linked Sias. Although many antibodies to Siglec-5 cross-react with Siglec-14, specific antibodies revealed that although Siglec-5 is expressed on neutrophils and B cells, Siglec-14 is found on neutrophils and monocytes. A SIGLEC14-null allele is frequently present in Asian populations but is less common in Europeans. The null allele arose from a recombination event between the 5′ region of the SIGLEC14 gene and the nearby 3′ region of the SIGLEC5 gene, resulting in a fusion protein almost identical to Siglec-5 but expressed in a Siglec-14-like manner. Individuals with chronic obstructive pulmonary disease (COPD) who are also SIGLEC14-null showed reduced exacerbation attacks (sudden worsening of symptoms) compared with individuals expressing Siglec-14. Both Siglec-5 and Siglec-14 can bind sialylated strains of Haemophilus influenzae implicated in COPD exacerbations and trigger inhibition and activation responses, respectively. Thus, the absence of Siglec-14 on neutrophils would lead to reduced inflammatory responses in SIGLEC14-null individuals with the added impact of inhibitory Siglec-5 on monocytes. Besides expression on leukocytes, both Siglec-5 and Siglec-14 are found on human amniotic epithelium and may mediate dualistic responses to GBS infection and the frequency of preterm births in infected mothers. There are no obvious equivalents of Siglec-5 or Siglec-14 in mice, making it difficult to study this interesting pair of receptors in vivo.
Siglec-6
Siglec-6 was cloned from a human placental cDNA library and was also independently identified during a screen for proteins that bind leptin, a hormone that regulates body weight. It has three Ig-like domains and the typical arrangement of ITIM and ITIM-like motifs in its cytoplasmic tail. Low levels of Siglec-6 are expressed on B cells, but high expression is seen in the placental cytotrophoblasts and syncytiotrophoblasts in humans. Siglec-6 levels are increased in preterm deliveries associated with preeclampsia, but it is not yet known if there is a causal relationship. The heavily sialylated protein glycodelin is produced in the uterus and appears to bind Siglec-6 on cytotrophoblasts and to suppress their migration into decidua through inhibition of ERK and c-Jun signaling. A SNP in SIGLEC6 is associated with SLE in Asians. Siglec-6 does not have an obvious ortholog in mice, but one is present in the chimpanzee and baboon. However, placental expression appears unique to humans.
Siglec-7, Siglec-9, and Siglec-E
Siglec-7 and Siglec-9 share a high degree of sequence similarity and appear to have evolved by gene duplication from an ancestral gene encoding a 3-Ig-domain inhibitory Siglec, represented in mice by Siglec-E. Siglec-7 is the major Siglec on human NK cells and is also seen at lower levels on monocytes, macrophages, DCs, and minor subsets of CD8 T cells. Disease-associated changes in expression patterns and levels of these Siglecs have been reported. Siglec-7 has also been detected in platelets, basophils, and mast cells in which it may modulate survival and activation. Siglec-9 is prominently expressed on neutrophils, monocytes, macrophages, and DCs, ∼30% of NK cells, and minor subsets of CD4 and CD8 T cells. Based on glycan array binding experiments (Chapter 29), Siglec-7 binds strongly to α2-8-linked Sias present in “b-series” gangliosides (Chapter 11) and some glycoproteins, whereas Siglec-9 prefers α2-3-linked Sias. Sulfation of the sialyl-Lex (SLex) structure can strongly influence recognition by both Siglecs, with Siglec-9 preferring 6-sulfo-SLex, and Siglec-7 binding well to both 6-sulfo-SLex and 6′-sulfo-SLex. Siglec-7 expression has also been reported in human pancreatic islet cell.
Siglec-E in mice shows a combination of some features of Siglec-7 and Siglec-9, being mainly expressed on neutrophils, monocytes, and macrophages, with Sia binding preferences that span those of both Siglec-7 and Siglec-9. Similar to T cells, NK cells in mice appear to lack expression of inhibitory Siglecs. Siglec-E is an important inhibitory receptor of neutrophils, as shown in multiple models such as LPS-induced lung inflammation in which Siglec-E-deficient mice show exaggerated CD11b-dependent neutrophil influx.
Tumor cells often up-regulate cell surface sialylated glycans and it appears that these may be important in Siglec-dependent dampening of antitumor immunity. Siglecs-7 and -9 can both suppress NK cell cytotoxicity against tumor cells expressing relevant glycan ligands. Siglec-9 and Siglec-E can also dampen neutrophil activation and tumor cell killing, whereas ligation of Siglec-9 or Siglec-E on macrophages by tumor-associated glycans seems to suppress formation of tumor-promoting M2 macrophages. In contrast to steady state, T cells in cancer patients express Siglec-9, and engagement of Siglec-9 by cancer-associated ligands may negatively impact tumor immunity, as do classic immune checkpoint receptor–ligand interactions. Overall Siglec effects on tumor cell biology may be dualistic, depending on the stage of tumor cell growth.
Studies with GBS have also shown that sialylated bacteria can subvert innate immune responses by targeting Siglec-9 or Siglec-E on neutrophils and macrophages, resulting in attenuation of phagocytosis, killing, and proinflammatory cytokine production. It has also been shown that myeloid Siglecs such as Siglec-9 and Siglec-E can modulate TLR signaling in response to pathogen ligands, leading to reduced secretion of proinflammatory mediators like TNF and increased production of anti-inflammatory cytokine, IL-10.
Siglec-8 and Siglec-F
Siglec-8 has three Ig domains and is expressed on eosinophils and mast cells, with weaker expression on basophils. It binds strongly to 6′-sulfo-SLex and to high-molecular-weight glycoproteins isolated from human airways. In mast cells, Siglec-8 ligation (with antibody) inhibits FcεRI-triggered degranulation responses, in line with its role as an inhibitory receptor. In eosinophils, Siglec-8 triggers apoptosis, which can occur following cross-linking with anti-Siglec-8 antibodies or sialoglycan polymers. Apoptosis depends on generation of reactive oxygen species and caspase activation, and is paradoxically enhanced in the presence of cytokine “survival” factors such as interleukin-5 (IL-5). The restricted expression of Siglec-8 on immune cells involved in allergy and the induction of cell death on antibody-mediated cross-linking make it an attractive therapeutic target for allergic diseases, and clinical trials of anti-Siglec-8 antibody in these diseases are ongoing.
Although there is no ortholog of Siglec-8 in mice, the four-Ig domain mouse Siglec-F is expressed in a similar way to Siglec-8 on eosinophils and has a similar glycan-binding preference for 6′-sulfo-SLex. It appears to have acquired similar functions through convergent evolution. There are some important differences, however. Siglec-F can recognize a broader range of α2-3-linked Sias, is also expressed on alveolar macrophages, and triggers weaker apoptosis using different signaling pathways.
Siglec-F-null mice show exaggerated eosinophilic responses in a lung allergy model, suggesting that its normal role is to dampen such responses. Interestingly, Siglec-F ligands in the airways and lung parenchyma were also up-regulated during allergic inflammation.
Siglec-10 and Siglec-G
Siglec-10 has five Ig-like domains, and in addition to the ITIM and ITIM-like motifs, displays an additional tyrosine-based motif (predicted to interact with Grb2) in its cytoplasmic tail. It is expressed at relatively low levels on several cells of the immune system, including B cells, monocytes, macrophages, and eosinophils. It can also be strongly up-regulated on tumor-infiltrating NK cells in hepatocellular carcinoma in which its expression was negatively associated with patient survival. It is the only CD33-related human Siglec that has a clear-cut ortholog in mice, designated Siglec-G. Both Siglec-10 and Siglec-G prefer Neu5Gc more than Neu5Ac in both α2-3 and α2-6 linkages. Similar to Siglec-10, Siglec-G is mainly expressed on B-cell subsets, DCs, and weakly on eosinophils. Mice deficient in Siglec-G show a 10-fold increase in numbers of a specialized subset of B lymphocytes, the B1a cells that make natural antibodies. These Siglec-G deficient B1a cells also show exaggerated Ca-fluxing following BCR cross-linking. Studies using “knock-in” mice carrying an inactivating mutation in the Sia-binding site of Siglec-G show a similar phenotype. This appears to be due to a requirement of Sia-dependent cis-interactions between Siglec-G and the BCR. On DCs, Siglec-G has been proposed to regulate cytokine responses to damage-associated molecular patterns (DAMPs) released by necrotic cells in sterile inflammation. This is thought to be due to a dampening effect of cis-interactions between Siglec-G and the heavily sialylated DAMP receptor, CD24. Disruption of this interaction through sialidases released by bacteria such as Streptococcus pneumoniae may be important in triggering inflammatory responses in sepsis. CD24 is overexpressed in some cancers and engages Siglec-10 on macrophages to suppress phagocytosis, in much the same way as does the classic “don't-eat-me” ligand–receptor pair, CD47 and SIRPα.
Siglec-11 and Siglec-16
Siglec-11 and Siglec-16 are paired inhibitory and activating receptors, with five and four Ig domains, respectively. In most humans, the SIGLEC16 gene has a 4-base-pair deletion, and only ∼35% of humans express one or two functional alleles. The extracellular regions of these proteins are >99% identical because of gene conversion events. Siglec-11 and Siglec-16 bind weakly to α2-8-linked Sias in vitro. These Siglecs appear to be absent from circulating leukocytes, but are expressed widely on populations of tissue macrophages, including resident microglia in the brain, where high levels of α2-8-linked Sias are present on gangliosides. Expression of Siglec-11 on microglia can impair their phagocytosis of apoptotic cells, yet alleviates neurotoxicity by suppressing neuroinflammation. Interestingly, microglial expression of Siglec-11 appears unique to humans. Siglec-16 is also present on microglia. These paired receptors exhibit dualistic responses to Escherichia coli K1, which expresses polysialic acid ligands.
HUMAN-SPECIFIC CHANGES IN SIGLEC BIOLOGY
The ancestral condition of some hominid Siglecs (e.g., Siglec-11) appears to have been preferential binding to Neu5Gc, a Sia specifically lost in human evolution ∼2–3 million years ago (Chapter 15). This loss could have resulted in some Siglec unmasking, possibly leading to a state of heightened innate immune reactivity. Some human Siglecs have undergone an adjustment to allow increased Neu5Ac binding, and the question arises whether the adjustment is yet complete. Possibly, as a consequence of Neu5Gc loss, several Siglecs seem to have undergone human-specific changes in comparison to our great ape evolutionary cousins. For example, Sn is expressed on most human macrophages, whereas only subsets of chimpanzee macrophages are positive. This may be related to the fact that Sn has a strong binding preference for Neu5Ac rather than Neu5Gc, which is not synthesized by humans. Human Siglec-5 and Siglec-14 appear to have undergone a restoration of the “essential” arginine residue needed for Sia recognition, which is mutated in chimpanzees, gorillas, and orangutans. Siglec-7 is selectively expressed in human pancreatic islets. The gene encoding Siglec-11 has undergone a human-specific gene conversion, resulting in a new protein expression in brain microglia along with its paired receptor, Siglec-16. Siglec-12 has suffered a human-specific and human universal inactivation of the “essential” arginine residue with subsequent permanent pseudogenization by a frameshift in some humans. Siglec-13 underwent a human-specific gene deletion and Siglec-17 has a frameshift mutation, events possibly occurring close to the time of the common ancestor of modern humans. Expression patterns of some Siglecs have also undergone changes, with placental expression of Siglec-6 and amniotic epithelium expression of Siglec-5/14 being human-specific, and a general suppression of all CD33rSiglecs on human T cells compared with the chimpanzee. The functional implications of these human-specific changes in Siglec biology for physiology and disease are under exploration and may be related to human propensities for certain diseases (Chapter 15).