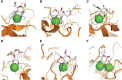The biological effects that glycans elicit are frequently dependent on recognition of specific glycan features by the proteins with which they interact. In this chapter, some of the key structural features underlying glycan–protein interactions, as well as the primary experimental methods that have led to an understanding of these features, are discussed, specifically X-ray crystallography, nuclear magnetic resonance (NMR), cryo-electron microscopy, and computational modeling.
BACKGROUND
As emphasized in previous chapters, the numbers of distinct glycans produced by various organisms are enormous, but at the same time, glycans lack the diversity in functional groups displayed by other molecules. To achieve specificity in glycan recognition, proteins rely as much on the stereospecific placement of glycan hydroxyl groups at chiral centers, use of different linkage sites, and extensive branching as they rely on specific modifications of hydroxyl groups by processes such as sulfation, phosphorylation, and esterification. This puts placement of various residues and functional groups in three dimensions at a premium. Building a three-dimensional picture of how recognition of glycans by proteins occurs is therefore essential if we are to understand how glycans are synthesized and recognized in the many physiological and pathological processes they control. It is also essential if we are to use knowledge of glycan recognition as a basis for the production of therapeutic agents that can control these processes in the event of disease. Building a structure depicting glycan recognition is not without its challenges. Most glycans are highly dynamic in solution, sampling many conformations. Often, a single or a small subset of conformations is selected when a complex forms. This works against the formation of stable complexes for structural studies and the direct use of solution conformational data in defining conformations of bound glycans.
The search for a structural basis of glycan recognition by proteins is not new. The concept of glycans fitting into pockets on protein surfaces dates back to Emil Fischer, who used the phrase “lock and key” to refer to enzymes that recognize specific glycan substrates. Lysozyme was the first “carbohydrate-binding protein” to be crystallized and have its three-dimensional structure determined. Subsequent work in the late 1960s and early 1970s led to a structure complexed with a tetrasaccharide that confirmed the existence of specific interactions occurring between sugars and proteins, and the ability of proteins to select the appropriate “key” from numerous possibilities.
Today, protein crystallography has reached a very high degree of sophistication and is responsible for the vast majority of the more than 170,000 structures deposited in the Protein Data Bank (PDB); however, producing a structure with ligands in place is still challenging. The structures that exist tend to have ligands that are relatively small and interact with particularly high binding constants. Glycan recognition frequently involves contacts with multiple residues to achieve specificity. So, native glycan ligands are often larger than other types of ligands. Often, high avidity is achieved through multivalent interactions, in which case the affinity for an isolated ligand–protein interaction is small. Nevertheless, there are a significant number of crystal structures for glycan–protein complexes, and these have contributed greatly to our understanding of the types of interactions that make glycan recognition possible.
Structural information on bound glycan ligands that is complementary to that from X-ray crystallography is increasingly coming from NMR methods. This is particularly valuable in that it is applicable to ligands with a broader range of affinities, including many that have the lower affinities amplified in multivalent interactions. It is also applicable in solution under near-physiological conditions in which concerns about the effects of crystal lattice contacts and occlusion of some interaction sites are absent. It is even possible to conduct some experiments on assemblies that mimic a membrane surface environment, an environment in which many protein–glycan interactions occur.
It is important to note that structural methodology is continually evolving, with additional information coming from techniques like small-angle X-ray scattering (SAXS) and cryo-electron microscopy (cryo-EM). Recent advances in cryo-EM provide many exciting opportunities to study protein–glycan interactions, which will also be discussed.
The fundamental understanding of glycan–protein interactions, as enriched by experimental studies of all types, has now been encoded in powerful molecular simulation programs that provide a computational approach to generating three dimensional pictures of glycan–protein complexes. These are important because it is difficult to produce complex glycan ligands in the amounts and purity required for most experimental approaches. These methods, although still evolving toward increased confidence in outcomes, provide models for experimentally inaccessible systems that can be tested with a variety of nonstructural approaches. They can also be leveraged with sparse structural data that alone could not provide detailed structural information.
CRYSTALLOGRAPHY
X-ray crystallography is a very powerful method for obtaining details of protein–ligand interactions. It excels in terms of the size range of molecules that can be studied (from small compounds to large multiprotein complexes) and in efficiency of data collection when high-energy X-ray beams at synchrotron sources are used. One of the limitations is still the crystallization step. Crystals of protein–carbohydrate complexes can be obtained by co-crystallizing the two partners or by soaking the carbohydrate ligand into an existing protein crystal. Because the quality of the crystal defines the limit of the diffraction pattern, and therefore the resolution of the structure, flexible oligosaccharide ligands may create structural heterogeneity and therefore limit the quality of the crystal. High-quality crystals of lectins are generally obtained with glycan ligands ranging from mono- to trisaccharides; glycosaminoglycan (GAG)-binding proteins or antibodies, which can bind much larger ligands, are more rarely crystallized in complex with carbohydrate ligands.
Diffraction data are now typically collected at very low temperatures, to protect molecules from radiation damage on high-energy synchrotron beam lines. Because freezing may damage the crystals owing to ice formation, glycerol is often used as cryoprotectant. Glycerol, with its carbohydrate-like hydroxylated carbons, is therefore frequently observed in glycan-binding sites, providing information about the amino acids involved in binding but sometimes competing with the carbohydrate ligand. Often, collaborative efforts with synthetic carbohydrate chemists are necessary to design, for example, nonhydrolyzable carbohydrate derivatives to obtain substrate and product-bound enzyme structures. These efforts can be combined with incorporating heavy atoms into the ligands, which in turn allow localizing them based on specific scattering characteristics.
Databases of Crystal Structures
Crystal structures of protein–carbohydrate complexes can be retrieved from different sources, including the PDB, but also from more specialized databases. The Carbohydrate-Active enZYmes (CAZY) database provides links to the PDB page for all crystal structures of glycosylhydrolases, glycosyltransferases, and their associated carbohydrate-binding modules. UniLectin3D is a database covering the three-dimensional features of lectins, and includes more than 2200 lectin three-dimensional structures, of which >60% are complexed with a carbohydrate ligand. The UniLectin3D classification of more than 500 distinct lectins results in 35 lectin domain folds, 109 classes, and 350 families sharing ∼20% and ∼70% sequence similarity, respectively. For each structure, links for coordinates, references, and taxonomy are provided, as well as glycan array data when available at the Consortium for Functional Glycomics. Mining for structural data is therefore possible, and structures can be analyzed at different levels revealing not only atomic details of the binding sites but also protein folds and oligomeric states. Examples are given below that illustrate how convergent evolution has built robust systems for efficient recognition of glycans by lectins.
Interactions in Carbohydrate-Binding Sites
The interactions between carbohydrates and amino acids include hydrogen bonds, van der Waals contacts, ionic bonds, and a number of more specialized interactions. CH-π interactions, for example, are associated with the frequent occurrence of aromatic amino acids in carbohydrate-binding sites. Water molecules are often observed that bridge between carbohydrate hydroxyl groups and amino acids. Interestingly, a significant number of enzymes and lectins use divalent ions that directly coordinate to the hydroxyl groups of carbohydrates and to side-chains of amino acids. Among the 350 different lectin families crystallized to date, more than 40 involve calcium ions in their binding sites. Most of them belong to the C-type lectin families (including selectins and DC-SIGN [dendritic cell–specific intercellular adhesion molecule-3-grabbing integrin]), but other types of lectins from different origins are also found to have one calcium ion in their binding site (Figure ). LecB from Pseudomonas aeruginosa requires the presence of two closely located calcium ions. Calcium ions contribute to the specificity of lectins by selecting for precise stereochemistries of hydroxyl groups; the two calcium ions of LecB, for example, only coordinate monosaccharides bearing the specific sequence of two equatorial and one axial hydroxyl group present in “fuco” and “manno” configurations. The ions also play a role in enhanced affinity through delocalization of charge as evaluated by quantum chemical calculations and through compensation for binding entropy losses by releasing strongly coordinated water molecules.
Graphical representation of six different calcium-dependent carbohydrate-binding sites found in crystal structures of lectins. (A) Human MBP-A complexed with mannoside (1KWU), (B) Pseudomonas aeruginosa LecA complexed with galactose (1OKO), (C) sea cucumber (more...)
Folding and Oligomerization Facilitate Binding to Cell Surfaces
Lectin structures adopt a limited number of folds (Figure ). Among them, there is a strong predominance of β-sheet-containing domains, such as β-sandwich, β-prism, β-trefoil, or β-propeller. The β-sandwich fold, which is an assembly of two β-sheets, characterizes a large family with different structures that vary in size and localization of binding sites. For example, fimbrial adhesins are very different from galectins in that they use a site near the edge of a sheet as opposed to the concave surface of a sheet. Some structural convergence is nevertheless observed. Intracellular animal lectins, which are involved in the quality control of glycoprotein synthesis, share the same protein fold with legume lectins.
(A) Distribution of the lectins with structures available in the Unilectin3D database as a function of fold family. (B) Graphical representation of some convergent β-propeller folds for lectins. The polypeptide chains are represented as ribbons (more...)
Convergence is also observed for the β-propeller fold, which is a circular arrangement of small β-sandwiches, called blades. Structures with five, six, or seven blades have been observed for lectins. With the exception of bacterial and fungal fucose-binding six-blade β-propellers, which are evolutionary related, these structures do not present sequence similarities. However, they share the same global shape allowing for the presentation of all binding sites on the same side of the “donut,” providing for very efficient multivalent binding to glycoconjugates on cell surfaces. This multivalent effect results in high avidity: PVL from the fungus Psathyrella velutina has an affinity of only 100 µm for GlcNAc at each binding site but an apparent avidity of 10 nm for GlcNAc presented on chips. This high avidity makes PVL an excellent tool for identifying tumor cells presenting truncated glycans with exposed GlcNAc.
NUCLEAR MAGNETIC RESONANCE
NMR can provide de novo high-resolution structures of proteins and glycan–protein complexes. It can also provide dynamic information when parts of bound glycans retain some of the mobility displayed in solution. However, NMR-based structure determination usually requires uniform isotopic labeling with magnetic nuclei such as 13C and 15N, to complement data from the highly abundant nucleus, 1H. Isotopic labeling can be accomplished when proteins can be expressed in bacterial hosts, but even then application is largely restricted to proteins of <20 kDa or of <40 kDa when perdeuteration can be used to improve resolution. The cost of uniform isotopic labeling often excludes application to many additional proteins of interest—in particular, glycoproteins—when expression in eukaryotic hosts proves essential. Hence, only a few complete structures of glycoproteins with native glycosylation have been produced by NMR methods. However, NMR has fewer restrictions when it builds on protein structures available from X-ray crystallography or computational modeling and capitalizes on its ability to focus on data involving actual glycan–protein interaction sites. We illustrate this potential in the following sections.
Chemical-Shift Mapping of Protein-Binding Sites for Glycans
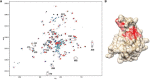
The initial step on the route to produce a three-dimensional structure of a protein by NMR methods is usually the assignment of backbone resonances, including the proton and nitrogen resonances of all amide 1H-15N pairs. This step is quite robust and can be accomplished in much less time, and on much larger targets, than a complete structure determination. These assignments are based on a series of multidimensional experiments that correlate chemical shifts of a series of directly bonded, NMR-active nuclear pairs. Among these is the two-dimensional 1H-15N heteronuclear single quantum coherence (HSQC) experiment, which correlates an amide 1H-15N pair through the appearance of a crosspeak at the chemical shifts of the amide proton and nitrogen of a particular protein residue. Once crosspeaks in this experiment are assigned, changes in chemical shift on addition of a glycan ligand can be used to identify a binding site. These changes often arise from small perturbations in residue geometry rather than a direct effect of the ligand on chemical shift, but the effects are usually sufficiently localized to identify the binding site. Figure shows an example of changes occurring on the interaction of a hexamer of chondroitin sulfate (CS), sulfated at the O4 position of each GalNAc residue (Chapters 3 and 17). There are actually two types of perturbations observed: gradual changes in chemical shift as ligand is added (arrows in Figure ) and the disappearance of one peak while another appears (ellipses in Figure ). These correspond to fast exchange on and off a weak binding site and slow exchange on and off a strong binding site, respectively. Perturbed residues can be mapped onto an existing structure of the protein, as shown in Figure for the strong binding site. As with many complexes involving a sulfated GAG, positively charged residues are involved; in this case histidine residues and a lysine residue are among those showing chemical shift changes. The advantage of these experiments is that a range of ligands can be examined, even those that may fail to produce well-ordered crystals for crystallographic analyses. A limitation is that the backbone resonances of the protein need to be assigned first.
Chemical shift mapping of slow and fast exchange binding sites for a 4-sulfated chondroitin sulfate (CS) hexamer on the Link module of TSG6. (A) Crosspeaks from spectra with increasing amounts of hexamer are superimposed. Those from residues experiencing (more...)
Identification of Bound Ligand Geometry and Ligand Interaction Surfaces
NMR also offers the potential for characterizing the geometry a ligand adopts on binding to a protein surface and the parts of the ligand that make contact with a protein. In both cases, the characterization stems from transfer of magnetization from one NMR active spin to another NMR active spin (usually protons) in a distance-dependent manner. In the case of bound ligand geometry, the experiment relies on a transferred nuclear Overhauser effect (trNOE). The basis is the same as for the NOE that is used in protein structure determination by NMR; however, as a large excess of ligand over protein is used (>10:1), only the ligand spectrum is observed. Measurements are usually made from crosspeaks in two-dimensional experiments similar to the HSQC experiment mentioned above, except that both dimensions are proton chemical shift, and crosspeaks have intensities dependent on the inverse sixth power of the distance between proton pairs (1/r6) rather than direct bonding. An average over both bound and free ligands is observed, but contributions are heavily weighted by those coming from the ligand in a complex because of scaling in proportion to molecular weight. This makes it possible to conduct trNOE experiments with a large excess of ligand and very little protein. Also, there is no requirement for isotopic labeling of either ligand or protein, and having a high-molecular-weight complex is an advantage. The geometry of the bound ligand is derived primarily from distances measured between protons that fall on opposite sides of a glycosidic bond. This distance then restrains glycosidic torsion angles accessible to structural models. Although there are many cases in which the bound geometry is similar to that of the dominant conformer found in solution, there are cases in which the geometry differs. Here, trNOE experiments offer unique insight that can guide synthesis of competitive inhibitors.
Transfer of magnetization from protons on a protein to protons on a ligand in an intermolecular NOE-like fashion can also provide information on the parts of a ligand in contact with amino acids in a protein's binding pocket (the ligand's interaction surface or binding epitope). In some cases, NOEs between a ligand proton and a specific amino acid proton can be observed, but this requires work with near-equimolar concentrations of ligand and protein, as well as full resonance assignment for both the ligand and the protein. A far more widely applied experiment sacrifices knowledge about specific protons on the protein for an ability to work with very large unlabeled and unassigned proteins. This experiment is called a saturation transfer difference (STD) NMR experiment. In fact, STD NMR investigations have also been conducted on some very large and complex systems including receptors embedded in membrane fragments, whole cells, and viruses. The experiment, which can be conducted at ratios of ligand to protein approaching 100:1, involves selective perturbation (saturation) of the magnetization of a set of protein protons and relies on the fact that the magnetization transfer between protons in large proteins is so efficient that it makes little difference where the change in magnetization is initiated; it can be from saturation of a methyl proton having a resonance at one extreme of the spectrum (upfield) or an aromatic proton having a resonance at the other extreme (downfield). Ideally, the saturation effect diffuses all over the protein protons and eventually is transferred to ligand protons close to the protein surface, and the resonances of these protons are reduced in intensity in a way that inversely correlates with the distances of a proton from the protein surface. Data are collected as a difference between one-dimensional proton spectra with and without saturation in the extremes of the protein spectrum. The resulting difference spectrum is dominated by resonances from the ligand that have contact with the protein. Mapping the position of protons assigned to these resonances onto a ligand structure allows depiction of the ligand binding epitope.
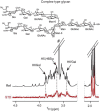
Figure shows an example that probes the interaction between a complex N-glycan (Chapters 3 and 9) and an HIV broadly neutralizing antibody. These antibodies specifically interact with surface glycans of HIV and are effective in inhibiting binding of the virus to target cells. Hence, there has been significant interest in exactly which glycans are recognized. Antibodies are large glycosylated proteins that are not usually amenable to NMR investigation by isotope-dependent methods, but STD NMR methods are applicable. The example uses a sample 20 µm in protein (Fab fragment) and 2 mm in glycan. Normal (reference) and STD NMR spectra are superimposed to show the saturated ligand resonances, which include some that come specifically from the Neu5Ac residues (Sia) on the termini of the glycan branches.
Binding epitope identification in a complex-type glycan bound to the HIV-1 neutralizing antibody PG16 using saturation transfer difference (STD) nuclear magnetic resonance (NMR) information. (Reproduced from Bewley CA, Shahzad-ul-Hussan S. 2013. Biopolymers (more...)
STD NMR applications involving long-chain multiantenna N-glycans, such as the one described above, are often hampered by the near-chemical equivalence of sites and degeneracy of resonances from the various branches of antenna. In these cases, the covalent attachment of lanthanide binding tags to the reducing end of the glycan has proven useful. The resulting dispersion of glycan signals due to pseudo-contact shifts can allow unambiguous determination of binding epitopes (see Further Reading).
Although the rapid dispersion of saturation throughout the protein in standard STD NMR experiments suppresses information about what protein residues are involved in binding, there are ways to retrieve some of this information. In multifrequency STD NMR spectra, as in the case of the differential-epitope-mapping method (DEEP-STD NMR), two STD NMR spectra are obtained using two very different saturating frequencies in spectral regions devoid of glycan signals—for example, one in the aromatic region of protein protons and the other in the aliphatic region. In each STD NMR spectrum, the ligand protons close to directly saturated protein protons (e.g., aromatic protons) show a little increase in STD intensity, in comparison to protons further away from the saturated protein protons. Analyzing and mapping those differences along the structure of the ligand (so-called differential epitope mapping) allows retrieval of information about which areas of the glycan interact with those different types of amino acids in the binding pocket. If the geometry of the pocket is known, this allows elucidation of the orientation or polarity of the glycan in the binding pocket.
The above provides a glimpse of NMR experiments that can be used to investigate protein–glycan interactions. There are many others that take advantage of additional properties such as differences in translational diffusion constants and specific interactions with water molecules. Many of these have been adopted as screening methods used in fragment based drug discovery programs. Information about these is available in Further Reading.
CRYO-ELECTRON MICROSCOPY
With the development of direct electron detectors, cryo-electron microscopy (cryo-EM) has become one of the most powerful techniques to obtain high-resolution structural information on biological macromolecules. With reported resolutions exceeding 2 Å, many molecular details are revealed under native-like conditions. By eliminating the need for growing well-ordered three-dimensional (3D) crystals and operating at fairly low sample concentrations, cryo-EM can provide atomic level insights into many biological samples, from soluble and membrane-integrated protein complexes to filamentous polymers and entire viruses.
Cryo-EM can be roughly divided into two main directions: one working with single (usually purified) particles, single-particle analysis (SPA), and the other using a tomography approach to analyze species in larger assemblies, such as in vitro–assembled scaffolds or even native cells and tissues.
For SPA, samples are generally analyzed in a thin layer of vitreous ice containing the particles in random orientations. Data is collected in the form of movies, which allows correction for beam-induced drift, followed by estimation of the contrast transfer function for each micrograph; both are necessary to obtain near-atomic resolutions. The individual particles are then computationally extracted from the micrographs, sorted, and ultimately aligned in three dimensions to reconstruct the molecular structure.
For cryo-electron tomography of, for example, a vitrified cell or virus particle, a tilt series of images is acquired to obtain different “specimen views” necessary for 3D reconstruction. This technique continues to face technical challenges, in part because of limitations on the tilt angles that can be achieved. However, it is a powerful tool to image, for example, the glycocalyx of various tissues, plant and fungal cells walls, or microbial cell envelopes and capsules.
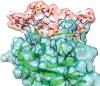
A major advantage of cryo-EM is that sample heterogeneity and/or conformational flexibility does not preclude analyses. To obtain well-diffracting crystals of a glycosylated protein, for example, the conformationally heterogenous glycans are often removed enzymatically to facilitate crystallization. For cryo-EM, these pretreatments are generally unnecessary, thereby providing molecular details of proteins in the context of posttranslational modifications. Analyses of fully glycosylated viral envelope proteins are fascinating examples that document the potential of cryo-EM for studying protein–carbohydrate interactions (Figure ). Other examples include polysaccharide-synthesizing enzymes bound to their polymeric products as well as integral membrane transporters associated with lipopolysaccharide substrates.
Cryo-electron microscopy structure of the native fully glycosylated HIV-1 envelope trimer. Protein subunits are shown as gray surfaces and glycans are shown as ball and sticks (PDB entry 5FUU). Carbohydrates are colored green and pale green (β- (more...)
We can thus look forward to correlating unprecedented structural insights on protein glycosylation and complex carbohydrate interactions with biochemical, functional, spectroscopic, and in silico approaches in the coming years.
COMPUTATIONAL MODELING
Experimental structural information obtained by crystallographic, NMR, and cryo-EM methods have clearly been of value in building an understanding of the molecular interactions that lead to glycan recognition by proteins. However, systems in which interactions are of interest far outnumber the cases in which these methods can be applied. Most crystal structures contain either small ligands or yield useful electron densities for only parts of larger ligands. The same is true to a certain extent for cryo-EM structures. NMR methods, although giving detailed information on bound ligand geometries, frequently give only qualitative information on parts of ligands or protein that are in intimate contact with each other. All three methods require substantial effort, particularly in preparing samples for investigation. A particular problem for glycans of interest is that they are often complex molecules that are difficult to prepare in highly pure forms or in the quantities needed for experimental investigation. There are also functionally important dynamic processes (e.g., enzymatic conversions of glycan substrates to products and transport of glycans) that are not well-represented by static, thermodynamically stable structures. Computational methods can extend analyses into these less accessible regions of structural investigation.
Computational Methods
Computational contributions to our understanding of glycan properties have a long history, beginning with a very fundamental understanding of factors influencing anomeric configuration and glycosidic torsion angles. These glycan-specific factors, such as the anomeric effect and the exo-anomeric effect, are described more thoroughly in Chapters 2, 3, and 50. When protein–glycan interactions are of interest, the situation becomes more complex with hydrogen bonding, van der Waals interactions, and electrostatic interactions between glycan and various amino acids becoming important. For very limited sets of atoms, it is possible to pursue an understanding of interactions using advanced quantum mechanical (QM) methods, but for larger systems other approaches based on semiempirical “force fields” are used, as in molecular mechanics (MM) and molecular dynamics (MD) simulations.
Empirical “force fields” used in MM and MD modules of packages such as Amber, CHARMM, and GROMOS are typically represented in terms of bond, bond angle, torsion angle, van der Waals, and electrostatic contributions to a molecular energy. Parameters in functions representing each of these terms have been optimized to reproduce QM as well as a selection of thermodynamic and spectroscopic data. Initially, these force fields were developed for proteins alone, so they did not include contributions such as the anomeric and exo-anomeric effects found in glycans. Subsequently, force fields explicitly designed to represent the energetics of glycans have been developed for use with these packages (e.g., the GLYCAM force field that is widely used with Amber). There still are challenges in simulating molecular interactions with these packages, among them perfecting models for solvent and accurately representing electrostatic interactions. These issues are very important for glycans, which are rich in hydroxyl groups that act as both hydrogen bond donors and acceptors in their interactions with water. Some glycans (e.g., GAGs) are highly charged, having both carboxylate groups and sulfate groups that interact strongly with positively charged amino acids in proteins and with water. Although early simulations were performed with implicit solvent models based on dielectric behavior, recent improvements in computational capabilities have allowed use of explicit solvent models, such as TIP3P and TIP5P.
MD, which uses the force fields directly in Newton's second law of motion, simulates movement of all atoms in addition to generating an ensemble of conformations and orientations that can be reached over times accessible to simulation (nsec to msec depending on the size of the system and efficiency of the computational platform). One advantage of MD is that certain important motional properties, such as the time for diffusion through a channel or the time needed for a conformational transition, can be modeled. One must remember, however, that force fields are meant to represent molecules near energy minima of a conformational surface and may not accurately represent the height of larger barriers separating different conformational states and certainly cannot represent changes in bonding that occur in a chemical reaction.
The actual characterization of how a ligand (a glycan in our case) interacts with a protein involves not just the conformational energetics of the free glycan, but also the conformational energetics of amino acid residues involved in the binding site and the energetics of the glycan–protein interaction. In some cases, there may be relatively little information on where the binding site on a protein is, so the characterization involves locating the best binding site, finding the best conformation for the ligand in the bound state, and finding the best conformations for the parts of the protein involved in binding. The whole process is referred to as “docking” a ligand onto a protein surface. Most docking programs (e.g., AutoDock, AutoDock Vina, and Glide) are designed to make the initial search for a site very efficient. To do this, they break the process into stages beginning with a rigid-body docking step that is designed to identify the best docking site and best initial “poses” for the ligand. Force fields are often simplified or interaction energies precalculated on a grid to speed up the process. Rigid-body docking generally works well for many small drug-like molecules. Also, in many situations, there is a crystal structure of the protein with a native ligand in the binding site, mitigating the problem of finding the binding pocket and optimizing side-chain conformations. For glycans, the situation is more complicated; the ligands are often flexible and protein structures with a native glycan in a binding site are often lacking.
In molecular docking, the objective is not to generate a single bound structure in the first stage but hundreds of “poses” that can be scored and ranked so that a subset can be selected for subsequent stages. Scoring functions are variable but usually include some sort of interaction energy as part of the score. Subsequent phases typically allow increased flexibility of side-chains and finally an MD refinement of poses, often in explicit water. Final scoring or ranking of poses by energy, even when performed with force fields used in MD programs, seldom leads to a single clear solution, and it has become common to filter poses with additional experimental information such as binding epitopes from STD NMR experiments or interactions with residues that have been identified as important in mutational studies.
Some docking programs are emerging (e.g., HADDOCK) that make use of experimental data in earlier stages to guide the selection of initial poses as well as maintain known preferences for glycan conformations or specific ligand–protein contacts. Some of the contributions to understanding of glycan–protein interactions that have come from docking exercises, as well as more advanced applications that merge QM with MD, are described in more detail in the following sections.
Docking of Heparan Sulfate Oligomers
Heparan sulfate (HS) chains, synthesized initially as a repeating disaccharide of glucuronic acid (GlcA) and N-acetylglucosamine (GlcNAc), and modified subsequently by sulfation and epimerization of some GlcA residues to iduronic acid (IdoA), are known to interact with a number of growth factors, receptors, and chemokines (Chapters 17 and 38). Despite the interest in the roles of these interactions in cell migration and differentiation, there are relatively few experimental structures depicting interactions with large HS fragments. The fact that suitable crystals are less apt to form in the presence of HS oligomers contributes to the lack of structures of complexes. Also, it is difficult to obtain homogeneous preparations of large oligomers, because of the variable sulfation patterns and variable conversion of GlcA to IdoA.
Computational modeling offers an alternative route to structures for many of these complexes. Specific patterns of sulfation and IdoA substitution are generated with ease. Yet, there are some challenges related to the flexibility of the HS chains and the ionic character of interactions that dominate their energetics; glycosidic angles in HS chains are variable, and IdoA rings sample several conformations, including a chair, 1C4, and a skew-boat conformer, 2S0. Moreover, orientations of the sulfate groups are variable, as are the side-chains of the lysine and arginine residues with which they tend to interact. Enhanced docking methodology combined with MD simulations overcomes some of these challenges.
Leukocyte common antigen-related protein (LAR) is a type IIa receptor protein tyrosine phosphatase (RPTP) important for signal transduction in biological processes, including axon growth and regeneration. Glycosaminoglycan chains, including HS, act as ligands that regulate LAR signal transduction. Knowing where HS binding sites are and what molecular interactions drive binding is an important step in the design of agents that could promote regeneration. Figure shows a snapshot of an HS pentamer bound to LAR. The structure was generated using the docking program HADDOCK. It employed several types of NMR data (chemical shift perturbation, STDs, and trNOEs) to guide selection of an initial set of 20 docked structures. The top scoring structures were subjected to short (50-nsec) MD runs in explicit water using GLYCAM06 force-field parameters for the HS fragment. The snapshot is from a longer (1-µsec) run, now highlighted in a movie (see Online Appendix 38A).
Docking of a heparan sulfate (HS) pentamer to the receptor protein tyrosine phosphatase, LAR (PDB entry 2YD5). (Docked structures as described in Gao N, et al. 2018. Biochemistry
57: 2189−2199.)
The interactions are typical of many GAG–protein interactions in that charged sulfates and carboxylates of the HS fragment interact with lysine and arginine residues of the protein binding site. These interactions are further stabilized by hydrophobic and hydrogen-bonding interactions with neighboring groups (e.g., with the glutamine residue in Figure ). The interactions with arginine are particularly important and, in addition to electrostatic contributions, often include those from bidentate hydrogen bonds between N-H groups on the arginine side-chain terminus and oxygens of sulfate groups. An example can be seen in the lower right where arginine 77 of the protein interacts with the N-sulfate on the terminal GlcN of the HS fragment.
Docking of Enzyme Substrates
A large number of enzymes are involved in the synthesis and degradation of glycans (more than 300 human enzymes). Their relative activities, combined with cellular location, are essential to the proper balance of these processes and any alteration, including genetic mutation, can lead to disease in humans. Pathogens also depend on similar processes, and understanding such mechanisms can facilitate the design of selective inhibitors of pathogen enzymes. This is another area in which molecular docking can play a role. Structural studies of glycan–protein complexes usually require a stable system—not one that would continually convert substrates to products. Molecular docking can provide useful depictions of these reactive systems.
A good example involves the glycosyltranferase, ST6Gal1. This is the enzyme that adds a sialic acid (typically Neu5Ac) to the galactose terminated branches of N-glycans by transferring Neu5Ac from its nucleotide-sugar donor, CMP-Neu5Ac, to an acceptor terminated with a Galβ1-4GlcNAc moiety (Chapter 6). The production of crystal structures of ST6Gal1, from both human and rat, opened the possibility of modeling at least a pretransition complex with both donor and acceptor in place. For the study discussed here, the crystal structure of the rat enzyme that contained neither donor nor acceptor (4MPS) was used as a starting point. The CMP-Neu5Ac was modeled into the active site based on the inactive donor analog in the crystal structure of the CstII protein (1RO7), which has a <20% sequence identity overall, but a much higher identity in the part of the active site that contains the donor. An initial structure for the minimal acceptor, Galβ1-4GlcNAc, was generated using the GLYCAM WebTool, but glycosidic bonds and hydroxyl groups were allowed to rotate during docking. Docking used the program AutoDock Vina. As in the previous example, an additional MD step in explicit water was used to refine the top ranked docked structure containing protein, donor, and acceptor. Interaction energies were then generated by applying MM/GBSA routines from Amber12 to 100-nsec MD production runs.
Although the positions of donor and amino acid residues near the donor were modeled to be quite similar to those seen in other transferases, the docking/MD procedure provides a unique view of a possible acceptor position and its interactions. Most of the interaction energy holding the acceptor in place comes from interactions with the galactose ring, which is well-positioned to allow nucleophilic attack on the anomeric carbon of the nucleotide-activated Neu5Ac. This energy results from hydrophobic stacking of Tyr-366 with the nonpolar face of the pyranose ring and a network of hydrogen bonds between Asp-271, Asn-230, His-367, and Gln-232 of the protein and O2, O3, O4, and O6 hydroxyl groups of Gal. The position of the GlcNAc is more variable but does contribute to binding energy. The position and interactions among protein, donor, and acceptor are depicted in Figure .
Stereo view of interactions between the donor (CMP-Neu5Ac), acceptor (GlcNAcβ1-4Gal), and protein residues in the active site of ST6Gal1. (Reproduced, with permission, from Meng L, et al. 2013. J Biol Chem
288: 34680−34698.)
FUTURE PROSPECTS
Structural biology is an evolving area of science both in terms of methodology and questions to be answered. The principle methodologies discussed here are each evolving: crystallographic methods using new X-ray sources (e.g., X-ray lasers) are allowing the analysis of microcrystals at room temperature and femtosecond timescales, thereby eliminating temperature- and beam-induced artifacts. Cryo-EM single-particle methods are approaching resolutions previously confined to X-ray crystallography. Single-particle and tomography EM approaches are continuing to undergo rapid development in terms of EM infrastructure, sample preparation, and data acquisition. Several user-friendly pipeline data processing packages exist, making this technology attractive to an increasing scientific audience. Hyperpolarization methods are reducing the sensitivity limitations of NMR, and solid-state NMR methods are allowing application to amorphous materials, including fibrils, cell-wall structures, and membrane fragments. Advances in computational technology are enabling simulation of ever larger systems and timescales. At the same time, structural targets are shifting from detailed characterization of single proteins and protein–glycan complexes to large-scale assemblies that cooperate to elicit a functional response. This is a promising situation for improved understanding of glycan function in biological systems.
ACKNOWLEDGMENTS
The authors appreciate helpful comments and suggestions from Barbara Mulloy, Dillon Chen, and Sean Stowell.
FURTHER READING
Pérez S, Tvaroska I. 2014. Carbohydrate–protein interactions: molecular modeling insights. In Advances in carbohydrate chemistry and biochemistry (ed. Baker DA, Horton D, editors. ), Vol. 71, pp. 9–136. Elsevier, Amsterdam. doi:10.1016/b978-0-12-800128-8.00001-7 [
PubMed: 25480504] [
CrossRef]
Bartesaghi A, Merk A, Banerjee S, Matthies D, Wu X, Milne JLS, Subramaniam S. 2015. 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science
348: 1147–1151. doi:10.2210/pdb5a1a/pdb [
PMC free article: PMC6512338] [
PubMed: 25953817] [
CrossRef]
Pomin VH, Mulloy B. 2015. Current structural biology of the heparin interactome. Curr Opin Struct Biol
34: 17–25. doi:10.1016/j.sbi.2015.05.007 [
PubMed: 26038285] [
CrossRef]
Canales A, Boos I, Perkams L, Karst L, Luber T, Karagiannis T, Domínguez G, Cañada FJ, Pérez-Castells J, Häusinger D, Unverzagt C, Jiménez-Barbero J. 2017. Breaking the limits in analyzing carbohydrate recognition by NMR spectroscopy: resolving branch-selective interaction of a tetra-antennary N-glycan with lectins. Angew Chem Int Ed
56: 14987–14991. doi:10.1002/ange.201709130 [
PMC free article: PMC5813150] [
PubMed: 28991403] [
CrossRef]
Glaeser RM. 2017. How good can cryo-EM become?
Nat Methods
13: 28–32. doi:10.1038/nmeth.3695 [
PubMed: 26716559] [
CrossRef]
Bonnardel F, Mariethoz J, Salentin S, Robin X, Schroeder M, Pérez S, Lisacek F, Imberty A. 2019. UniLectin3D, a database of carbohydrate binding proteins with curated information on 3D structures and interacting ligands. Nucleic Acid Res
47: D1236–D1244. doi:10.1093/nar/gky832 [
PMC free article: PMC6323968] [
PubMed: 30239928] [
CrossRef]