Summary
Clinical characteristics.
Childhood ataxia with central nervous system hypomyelination / vanishing white matter (CACH/VWM) is characterized by ataxia, spasticity, and variable optic atrophy. The phenotypic range includes a prenatal/congenital form, a subacute infantile form (onset age <1 year), an early childhood-onset form (onset age 1 to <4 years), a late childhood-/juvenile-onset form (onset age 4 to <18 years), and an adult-onset form (onset ≥18 years). The prenatal/congenital form is characterized by severe encephalopathy. In the later-onset forms initial motor and intellectual development is normal or mildly delayed, followed by neurologic deterioration with a chronic progressive or subacute course. While in childhood-onset forms motor deterioration dominates, in adult-onset forms cognitive decline and personality changes dominate. Chronic progressive decline can be exacerbated by rapid deterioration during febrile illnesses or following head trauma or major surgical procedures, or by acute and extreme fright.
Diagnosis/testing.
The diagnosis of CACH/VWM can be established in an individual with typical clinical findings, characteristic abnormalities on cranial MRI, and identification of biallelic pathogenic variants in one of five genes (EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5), which encode the five subunits of the eukaryotic translation initiation factor 2B (eIF2B).
Management.
Treatment of manifestations: Physical therapy and rehabilitation for motor dysfunction (mainly spasticity and ataxia); anti-seizure medication for seizures.
Prevention of secondary complications: Prevention of infections and fever when possible through the use of vaccinations, low-dose maintenance antibiotics during winter, antibiotics for minor infections, and antipyretics for fever. For children, wearing a helmet when outside helps minimize the effects of head trauma.
Surveillance: Close monitoring of neurologic status for several days during febrile infections and following head trauma or surgical procedures with anesthesia.
Agents/circumstances to avoid: Contact sports, head trauma, infections, high body temperature and, if possible, major surgery.
Genetic counseling.
CACH/VWM is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Prenatal diagnosis for pregnancies at increased risk is possible if the pathogenic variants in an affected relative have been identified.
Diagnosis
Suggestive Findings
Childhood ataxia with central nervous system hypomyelination / vanishing white matter (CACH/VWM) should be suspected in individuals with the following clinical, laboratory, and imaging findings.
Clinical findings
- Antenatal/early-infantile form:
- Oligohydramnios
- Intrauterine growth retardation
- Severe encephalopathy
- Microcephaly
- Contractures
- Cataract
- Pancreatitis
- Hepatosplenomegaly
- Renal hypoplasia
- Later-onset form:
- Initial motor and intellectual development is normal or mildly delayed.
- Neurologic deterioration has a chronic progressive or subacute course. Episodes of subacute deterioration may follow minor infection or minor head trauma and may lead to lethargy or coma.
- Truncal and appendicular ataxia
- Spasticity with increased tendon reflexes
- Peripheral nervous system is usually not involved.
- Optic atrophy may develop.
- Epilepsy may occur but is not the predominant sign of the disease except in an acute situation.
- In children, intellectual abilities may be affected but not to the same degree as motor functions. Alteration in intellectual abilities associated with behavioral changes can be the initial symptom in adult-onset forms.
- Ovarian failure may be present as primary or secondary amenorrhea [Hamilton et al 2018].
Laboratory findings [van der Knaap et al 1999]
- Routine cerebrospinal fluid (CSF) analysis is normal.
- Glycine is often elevated.
MRI findings
- The cerebral hemispheric white matter is symmetrically and diffusely abnormal.
- On T1-weighted and FLAIR images, a fine meshwork of remaining tissue strands is usually visible within the areas of CSF-like white matter, with a typical radiating appearance on sagittal and coronal images and a dot-like pattern in the centrum semiovale on the transverse images (Figure 4) [van der Knaap et al 2006].
- The MRI abnormalities are present in all affected individuals regardless of age of onset and are even present in asymptomatic affected sibs of a proband, although in presymptomatic and early symptomatic individuals the cerebral white matter may be abnormal on MRI, but not yet CSF-like. Over time, increasing amounts of white matter vanish and are replaced with CSF; cystic breakdown of the white matter is seen on proton density or FLAIR images [van der Knaap et al 2006]. Cerebellar atrophy varies from mild to severe and primarily involves the vermis.
- Severe cerebral atrophy can be observed in adult-onset forms with slow progression. Cranial CT scan is of limited use and usually shows diffuse and symmetric hypodensity of the cerebral hemispheric white matter with no calcifications.
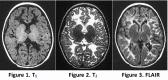
Figure
MRI of an individual with the classic form of CACH/VWM Figure 1. Diffuse hypointensity of the white matter on T1-weighted images
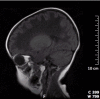
Figure 4.
Parasagittal T1-weighted MRI image of an individual with CACH/VWM shows diffuse hypointensity of the white matter interrupted by a typical meshwork of remaining tissue strands radiating across the abnormal white matter.
Establishing the Diagnosis
The diagnosis of CACH/VWM is established in a proband with the above Suggestive Findings and/or biallelic pathogenic variants in one of the genes listed in Table 1 identified on molecular genetic testing.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of CACH/VWM is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of CACH/VWM has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of CACH/VWM, molecular genetic testing approaches can include use of a multigene panel; a panel that includes the genes listed in Table 1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the diagnosis of CACH/VWM is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Childhood Ataxia with Central Nervous System Hypomyelination / Vanishing White Matter (CACH/VWM)
Incidental findings*
- Eukaryotic translation initiation factor 2B (eIF2B) guanine exchange factor (GEF) activity measured in lymphoblastoid cell lines from affected individuals was found to be lower in most persons with pathogenic variants in EIF2B1, EIF2B2, EIF2B3, EIF2B4, or EIF2B5 than in control subjects [Fogli et al 2004b]. eIF2B GEF activity assays in lymphoblastoid cell lines from 63 affected persons presenting with different clinical forms and pathogenic variants EIF2B1, EIF2B2, EIF2B3, EIF2B4, or EIF2B5 showed significantly decreased GEF activity with 100% specificity and 89% sensitivity when the activity threshold was set at 77.5% of normal [Horzinski et al 2009]. In the early-infantile form of the disease (onset age <1 year) the GEF activity was below the threshold of 77.5% of normal. Persons with late-onset disease and a wide variety of pathogenic variants (Table 1) had higher GEF activity that overlapped with the normal range. A significant decrease of GEF activity has also been reported in the 8/8 EIF2B1-5-mutated lymphoblastoid cell lines and 3/4 fibroblast cell lines analyzed by Liu et al [2011]. However, no correlation between eIF2B GEF activity and disease severity was found in this study. The findings were substantiated by similar results in transfected HEK293 cells [Liu et al 2011]. Thus, it can be concluded that if decreased activity is found, CACH/VWM is the most likely diagnosis; but if normal or increased activity is found, CACH/VWM cannot be ruled out.
- The CSF glycine level was found to be elevated in persons with CACH/VWM, in whom the diagnosis was later molecularly confirmed [van der Knaap et al 1999]. This test is not specific and its sensitivity is not known.
- The CSF asialotransferrin/total transferrin ratio was found to be low in persons with genetically confirmed CACH/VWM, a finding that can help identify those likely to have pathogenic variants in any of the five genes encoding the eIF2B subunits detected on sequence analysis [Vanderver et al 2005]. Note: This test is cumbersome and not generally available.
* Testing is on a research basis only.
Clinical Characteristics
Clinical Description
Childhood ataxia with central nervous system hypomyelination / vanishing white matter (CACH/VWM) phenotypes range from a congenital or early-infantile form (onset age <1 year) to an early childhood-onset form (onset age 1 to <4 years), a late-childhood/juvenile-onset form (onset age 4 to <18 years), and an adult-onset form (onset ≥18 years [Hamilton et al 2018]. Both the childhood and juvenile forms have been observed in sibs; the infantile and juvenile/adult forms have never been observed within the same family [Hamilton et al 2018].
Neurology. The neurologic signs depend on the age of onset [Hamilton et al 2018]. In the congenital and early-infantile forms, the encephalopathy is severe, seizures are often a predominant clinical feature, and decline is rapid and followed quickly by death. In early and late childhood-onset forms, motor decline predominates with ataxia and spasticity. Cognitive decline is relatively mild. Adult-onset forms usually present with cognitive decline and mood and personality changes; motor decline comes later in the disease course. Optic atrophy is variable and rather late in all forms.
The rate of disease progression depends on the age of onset. For individuals with disease onset before age four years, the decline is in general more rapid and more severe the earlier the onset. For onset after age four years, the disease course is generally slower and milder and life span is longer. For this later-onset group, however, variation in severity is wide and does not correlate with specific age at onset [Hamilton et al 2018]. Chronic progressive decline can be exacerbated by rapid deterioration during febrile illness or following minor head trauma or fright; such exacerbations are more common in early-onset than in later-onset forms of the disease [Hamilton et al 2018]. Rarely, a child or an adult with normal neurologic examination and biallelic pathogenic variants in one of the genes listed in Table 1 is identified by brain MRI that is performed because of headache or dizziness [Fontenelle et al 2008]. Follow up of such individuals for more than ten years may show no neurologic deterioration [Hamilton et al 2018; R Schiffmann, unpublished data].
Ovarian failure. While the juvenile and adult forms are often associated with primary or secondary ovarian failure – a syndrome referred to as "ovarioleukodystrophy" [Schiffmann et al 1997, Fogli et al 2003], ovarian failure may occur in any of the forms regardless of age of onset; it has been found at autopsy in infantile and childhood cases [van der Knaap et al 2003]. Because the affected individuals were prepubertal, the ovarian dysgenesis was not clinically manifest.
Antenatal form. The antenatal-onset form presents in the third trimester of pregnancy with oligohydramnios and decreased fetal movement [van der Knaap et al 2003, Hamilton et al 2018]. Clinical features that may be noted soon after birth include feeding difficulties, vomiting, hypotonia, mild contractures, cataract (sometimes oil droplet cataract), and microcephaly. Apathy, intractable seizures, and finally apneic spells and coma follow. Other organ involvement can include hepatosplenomegaly, renal hypoplasia, pancreatitis, and ovarian dysgenesis.
The clinical course is rapidly and relentlessly downhill; the adverse effect of stress factors is less clear. So far, all infants with neonatal presentation have died within the first year of life [Hamilton et al 2018].
Infantile form. A rapidly fatal severe form of CACH/VWM is characterized by onset in the first year of life and death a few months later [Fogli et al 2002, Hamilton et al 2018]. Affected infants develop irritability, stupor, and rapid loss of motor abilities, with or without a preceding intercurrent infection.
A specific infantile-onset phenotype was described as "Cree leukoencephalopathy" because of its occurrence in the native North American Cree and Chippewayan indigenous population [Fogli et al 2002]. Infants typically have hypotonia followed by sudden onset of seizures (age 3-6 months), spasticity, vomiting (often with fever), developmental regression, blindness, lethargy, and cessation of head growth, with death by age two years.
Early childhood-onset form. Initially most children develop normally; some have mild motor or speech delay. New-onset ataxia is the most common initial symptom between ages one and four years. Some children develop dysmetric tremor or become comatose spontaneously or acutely following mild head trauma or febrile illness [Hamilton et al 2018].
Subsequently, generally progressive deterioration results in increasing difficulty in walking, tremor, spasticity with hyperreflexia, dysarthria, and seizures. Once a child becomes nonambulatory, the clinical course may remain stable for several years. Swallowing difficulties and optic atrophy develop late in the disease.
Head circumference is usually normal; however, severe progressive macrocephaly occurring after age two years has been reported [Hamilton et al 2018]; microcephaly has also been observed. The peripheral nervous system is usually normal, although predominantly sensory nerve involvement has been reported [Federico et al 2006, Huntsman et al 2007]. Intellectual abilities are relatively preserved.
The time course of disease progression varies among individuals even within the same family, ranging from rapid progression with death occurring one to five years after onset to very slow progression with death occurring decades after onset.
Late childhood/juvenile-onset form. Children develop symptoms between ages four and 18 years. They often have a slowly progressive spastic diplegia, relative sparing of cognitive ability, and likely long-term survival with long periods of stability and even temporary improvement of motor function [Hamilton et al 2018]. However, the disease course is highly variable and rapid progression and death after a few months have also been described [Hamilton et al 2018].
Adult-onset form. Behavioral problems associated with cognitive decline are frequently reported before neurologic symptoms appear [Labauge et al 2009, Hamilton et al 2018]. Acute, transient neurologic symptoms (optic neuritis, hemiparesis) or severe headache – as well as primary or secondary amenorrhea in females – can be the presenting symptoms.
Asymptomatic and minimally symptomatic adults with two pathogenic variants in one of the genes and a typically affected sib have also been described [Leegwater et al 2001, Hamilton et al 2018].
Neuropathologic findings in general are a cavitating orthochromatic leukodystrophy with rarity of myelin breakdown and relative sparing of axons [Bugiani et al 2010]. Vacuolization and cavitation of the white matter are diffuse, giving a spongiform appearance. Cerebral and cerebellar myelin is markedly diminished. The spinal cord is also affected [Leferink et al 2018]. Oligodendrocytes are increased in number, whereas astrogliosis is feeble [Bugiani et al 2011]. The hallmark is the presence of oligodendrocytes with "foamy" cytoplasm and markedly abnormal astrocytes with few stunted processes [Wong et al 2000, Bugiani et al 2011]. The white matter astrocytes and oligodendrocytes are immature and are, in fact, astrocyte and oligodendrocyte precursor cells, explaining the lack of myelin production and scarce gliosis [Bugiani et al 2011, Bugiani et al 2013].
Phenotype Correlations by Gene
No statistically significant differences have been observed for individuals with pathogenic variants in EIF2B1, EIF2B2, EIF2B3, EIF2B4, or EIF2B5 regarding age of onset and survival [Hamilton et al 2018]. Only for the parameter "loss of walking without support," an overall significant difference was observed: ambulation was better preserved in individuals with EIF1B1 pathogenic variants, but this group was very small (only five individuals) and therefore not necessarily representative; when excluding these five individuals from the analysis, no overall differences remained for individuals with pathogenic variants in EIF2B2, EIF2B3, EIF2B4, or EIF2B5 [Hamilton et al 2018].
Genotype-Phenotype Correlations
Although intrafamilial variability exists, correlation between certain homozygous pathogenic variants and age of onset and disease severity has been described [Fogli et al 2004a, van der Lei et al 2010, Hamilton et al 2018]. A recent study of 296 individuals with CACH/VWM compared all available groups of at least three affected individuals from different families with the same pathogenic variants. In most groups with a similar genotype, severity measures, such as age of onset and survival, were rather consistent, but some variability was present, especially for pathogenic variants associated with a milder phenotype.
EIF2B5
- In individuals homozygous for the p.Thr91Ala pathogenic variant, the phenotype may vary from childhood onset to adults with no symptoms [Hamilton et al 2018].
- Certain EIF2B5 homozygous pathogenic variants, such as p.Arg113His, are most often (though not invariably) associated with a mild disease form and never give rise to the infantile type [Hamilton et al 2018].
- Certain EIF2B5 pathogenic variants, such as those at p.Arg339 (p.Arg339Trp, p.Arg339Gln, or p.Arg339Pro) and p.Val309Leu, are predictably associated with severe disease [Leegwater et al 2001, Fogli et al 2004a, van der Lei et al 2010].
Penetrance
Some adults who are homozygous or compound heterozygous for two disease-causing pathogenic variants in the same gene may be asymptomatic for prolonged periods of time [Hamilton et al 2018].
Nomenclature
"Cree leukoencephalopathy," described in the native North American Cree and Chippewayan indigenous population, is now recognized to be an infantile form of CACH/VWM [Fogli et al 2002].
Prevalence
The prevalence of CACH/VWM is not known; it is considered one of the more common leukodystrophies. In the Netherlands, the live-birth incidence was recently shown to be 1:80,000 [Hamilton et al 2018] and the prevalence of known, living affected individuals is 1.4:1,000,000.
Genetically Related (Allelic) Disorders
Thus far, all individuals with eIF2B-related disease have a leukodystrophy; no other phenotypes have been observed.
A contiguous gene deletion that included EIF2B2 and an additional heterozygous EIF2B2 pathogenic variant were reported in a child with features of CACH/VWM plus dysmorphic facial features [Shimada et al 2012].
Differential Diagnosis
Table 2.
Other Disorders Affecting the White Matter Diffusely During Childhood to Consider in the Differential Diagnosis of CACH/VWM
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with childhood ataxia with central nervous system hypomyelination / vanishing white matter (CACH/VWM), the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Brain MRI
- Ophthalmologic examination
- Neurologic examination
- Physical therapy/occupational therapy assessment as needed
- Consultation with a clinical geneticist and/or genetic counselor
Note: If an individual is diagnosed while asymptomatic, either because of an affected sib or as an incidental finding on exome sequencing, the above evaluations and the recommendations in Prevention of Secondary Complications should be applied.
Treatment of Manifestations
The following are appropriate:
- Physical therapy and rehabilitation for motor dysfunction (mainly spasticity and ataxia)
- Ankle-foot orthotics in individuals with hypotonia and weakness of ankle dorsiflexors
- Anti-seizure medication for treatment of seizures and abnormalities of behavior and mood
Prevention of Secondary Complications
Considering the known adverse effect of fever, it is important to prevent infections and fever as much as possible (e.g., through the use of vaccinations, including anti-flu vaccination); low-dose maintenance antibiotics during winter time, antibiotics for minor infections, and antipyretics for fever are appropriate. For children, wearing a helmet while outside helps minimize the effects of possible head trauma.
Surveillance
Close surveillance for several days following head trauma or major surgical procedure with anesthesia is indicated because neurologic deterioration (presumably stress related) may follow.
Agents/Circumstances to Avoid
Avoid the following:
- Contact sports and other activities with a high risk of head trauma
- Stressful emotional and physical situations (e.g., acute fright, fever and other causes of extreme temperatures, major surgery)
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Other
In general, corticosteriods and intravenous gamma globulin are not effective in the treatment of CACH/VWM. Corticosteriods have been used with inconsistent results in acute situations, including intractable status epilepticus.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Childhood ataxia with central nervous system hypomyelination / vanishing white matter (CACH/VWM) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one CACH/VWM-causing pathogenic variant).
- Heterozygotes (carriers) are asymptomatic. No clinical or MRI abnormalities have been found in carriers of pathogenic variants in EIF2B1, EIF2B2, EIF2B3, EIF2B4, or EIF2B5.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Age of onset of neurologic signs can differ from one individual to another within the same family. Therefore, a neurologically asymptomatic sib of an affected individual may have biallelic pathogenic variants and be at high risk of developing the disease. The large majority of (if not all) apparently asymptomatic individuals appear to have the extensive white matter abnormalities characteristic of the syndrome on head MRI, and may have very mild learning, cognitive, or behavioral disabilities. Note: Although both the childhood and juvenile forms have been observed in sibs [Leegwater et al 2001], the infantile and juvenile/adult forms have never been observed within the same family [Hamilton et al 2018.
- Heterozygotes (carriers) are asymptomatic. No clinical or MRI abnormalities have been found in carriers of a CACH/VWM-causing pathogenic variant.
Offspring of a proband. The offspring of an individual with CACH/VWM are obligate heterozygotes (carriers) for a CACH/VWM-causing pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a CACH/VWM-causing pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the CACH/VWM-causing pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to parents of an affected individual and to young adults who are affected or at risk, or are carriers.
Predictive testing (i.e., testing of asymptomatic at-risk individuals)
- Predictive testing for at-risk individuals is possible once the CACH/VWM-causing pathogenic variants have been identified in an affected family member.
- Potential consequences of such testing (including but not limited to socioeconomic changes and the need for long-term follow up and evaluation arrangements for individuals with a positive test result) as well as the capabilities and limitations of predictive testing should be discussed in the context of formal genetic counseling prior to testing.
- Predictive testing is not useful in predicting age of onset, severity, type of symptoms, or rate of progression in asymptomatic individuals.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown).
Prenatal Testing and Preimplantation Genetic Testing
Once the CACH/VWM-causing pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- National Library of Medicine Genetics Home Reference
- European Leukodystrophy Association (ELA)Phone: 03 83 30 93 34
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- United Leukodystrophy FoundationPhone: 800-SAV-LIVE; 815-748-3211Email: [email protected]
- Myelin Disorders Bioregistry ProjectPhone: 215-590-1719Email: [email protected]
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Childhood Ataxia with Central Nervous System Hypomyelination / Vanishing White Matter: Genes and Databases
Table B.
OMIM Entries for Childhood Ataxia with Central Nervous System Hypomyelination / Vanishing White Matter (View All in OMIM)
Molecular Pathogenesis
Introduction. The eukaryotic translation initiation factor eIF2B is composed of five subunits. Its function is to convert protein synthesis initiation factor 2 (eIF2) from an inactive GDP-bound form to an active eIF2-GTP complex, allowing the formation of the 43S complex, needed for initiation of mRNA translation. It is not yet understood why a defect in eIF2B, a ubiquitous protein complex, affects predominantly the brain white matter. The crucial role of eIF2B as regulator of protein synthesis under stress conditions could explain the neurologic deterioration during or after head trauma and fever [Scheper et al 2006].
The biologic importance of the eIF2B complex is evidenced by the following:
- Yeast with null pathogenic variants for any of the five genes EIF2B1-EIF2B5 is not viable.
- Pathogenic variants that completely abolish eIF2B activity are probably lethal in the biallellic state in humans; nonsense variants are rare and only observed in compound heterozygotes in association with a pathogenic missense variant [van der Knaap et al 2010].
- Affected individuals have decreased GEF activity (20%-77% of normal) [Fogli et al 2004b, Horzinski et al 2009].
- Pathogenic variants in EIF2B1-EIF2B5 that decrease guanine exchange factor (GEF) activity result from aberrant protein folding, leading to an impaired ability to form functional eIF2B complexes that bind substrate normally [Li et al 2004, Richardson et al 2004].
Of note, decreased GEF activity leads to enhanced translation of specific mRNA of proteins, similar to the situation that occurs when a cell is under stress.
Mechanism of disease causation. All disease-associated variants are loss-of-function variants, hampering eIF2B activity, thus impairing cell stress responses.
Table 3.
Childhood Ataxia with Central Nervous System Hypomyelination / Vanishing White Matter: Notable Pathogenic Variants by Gene
References
Literature Cited
- Alamri H, Al Mutairi F, Alothman J, Alothaim A, Alfadhel M, Alfares A. Diabetic ketoacidosis in vanishing white matter. Clin Case Rep. 2016;4:717–20. [PMC free article: PMC4974412] [PubMed: 27525068]
- Bugiani M, Boor I, Powers JM, Scheper GC, van der Knaap MS. Leukoencephalopathy with vanishing white matter: a review. J Neuropathol Exp Neurol. 2010;69:987–96. [PubMed: 20838246]
- Bugiani M, Boor I, van Kollenburg B, Postma N, Polder E, van Berkel C, van Kesteren RE, Windrem MS, Hol EM, Scheper GC, Goldman SA, van der Knaap MS. Defective glial maturation in vanishing white matter disease. J Neuropathol Exp Neurol. 2011;70:69–82. [PMC free article: PMC4135437] [PubMed: 21157376]
- Bugiani M, Postma N, Polder E, Dieleman N, Scheffer PG, Sim FJ, van der Knaap MS, Boor I. Hyaluronan accumulation and arrested oligodendrocyte progenitor maturation in vanishing white matter disease. Brain. 2013;136:209–22. [PubMed: 23365098]
- Federico A, Scali O, Stromillo ML, Di Perri C, Bianchi S, Sicurelli F, De Stefano N, Malandrini A, Dotti MT. Peripheral neuropathy in vanishing white matter disease with a novel EIF2B5 mutation. Neurology. 2006;67:353–5. [PubMed: 16864840]
- Fogli A, Rodriguez D, Eymard-Pierre E, Bouhour F, Labauge P, Meaney BF, Zeesman S, Kaneski CR, Schiffmann R, Boespflug-Tanguy O. Ovarian failure related to eukaryotic initiation factor 2B mutations. Am J Hum Genet. 2003;72:1544–50. [PMC free article: PMC1180314] [PubMed: 12707859]
- Fogli A, Schiffmann R, Bertini E, Ughetto S, Combes P, Eymard-Pierre E, Kaneski CR, Pineda M, Troncoso M, Uziel G, Surtees R, Pugin D, Chaunu MP, Rodriguez D, Boespflug-Tanguy O. The effect of genotype on the natural history of eIF2B-related leukodystrophies. Neurology. 2004a;62:1509–17. [PubMed: 15136673]
- Fogli A, Schiffmann R, Hugendubler L, Combes P, Bertini E, Rodriguez D, Kimball SR, Boespflug-Tanguy O. Decreased guanine nucleotide exchange factor activity in eIF2B-mutated patients. Eur J Hum Genet. 2004b;12:561–6. [PubMed: 15054402]
- Fogli A, Wong K, Eymard-Pierre E, Wenger J, Bouffard JP, Goldin E, Black DN, Boespflug-Tanguy O, Schiffmann R. Cree leukoencephalopathy and CACH/VWM disease are allelic at the EIF2B5 locus. Ann Neurol. 2002;52:506–10. [PubMed: 12325082]
- Fontenelle LM, Scheper GC, Brandao L, van der Knaap MS. Atypical presentation of vanishing white matter disease. Arq Neuropsiquiatr. 2008;66:549–51. [PubMed: 18813718]
- Gowda VK, Srinivasan VM, Bhat M, Benakappa A. Case of childhood ataxia with central nervous system hypomyelination with a novel mutation in EIF2B3 gene. J Pediatr Neurosci. 2017;12:196–8. [PMC free article: PMC5588653] [PubMed: 28904586]
- Hamilton EMC, van der Lei HDW, Vermeulen G, Gerver JAM, Lourenço CM, Naidu S, Mierzewska H, Gemke RJBJ, de Vet HCW, Uitdehaag BMJ, Lissenberg-Witte BI, van der Knaap MS, et al. The natural history of vanishing white matter. Ann Neurol. 2018;84:274–88. [PMC free article: PMC6175238] [PubMed: 30014503]
- Hettiaracchchi D, Neththikumara N, Pathirana BAPS, Padeniya A, Dissanayake VHW. A novel mutation in the EIF2B4 gene associated with leukoencephalopathy with vanishing white matter. Case Rep Pediatr. 2018;2018:2731039. [PMC free article: PMC6057309] [PubMed: 30073106]
- Horzinski L, Huyghe A, Cardoso MC, Gonthier C, Ouchchane L, Schiffmann R, Blanc P, Boespflug-Tanguy O, Fogli A. Eukaryotic initiation factor 2B (eIF2B) GEF activity as a diagnostic tool for EIF2B-related disorders. PLoS One. 2009;4:e8318. [PMC free article: PMC2789406] [PubMed: 20016818]
- Huntsman RJ, Seshia S, Lowry N, Lemire EG, Harder SL. Peripheral neuropathy in a child with Cree leukodystrophy. J Child Neurol. 2007;22:766–8. [PubMed: 17641267]
- Kanbayashi T, Saito F, Matsukawa T, Oba H, Hokkoku K, Hatanaka Y, Tsuji S, Sonoo M. Adult-onset vanishing white matter disease with novel missense mutations in a subunit of translational regulator, EIF2B4. Clin Genet. 2015;88:401–3. [PubMed: 25600065]
- Labauge P, Horzinski L, Ayrignac X, Blanc P, Vukusic S, Rodriguez D, Mauguiere F, Peter L, Goizet C, Bouhour F, Denier C, Confavreux C, Obadia M, Blanc F, de Sèze J, Fogli A, Boespflug-Tanguy O. Natural history of adult-onset eIF2B-related disorders: a multi-centric survey of 16 cases. Brain. 2009;132:2161–9. [PubMed: 19625339]
- Leegwater PA, Vermeulen G, Konst AA, Naidu S, Mulders J, Visser A, Kersbergen P, Mobach D, Fonds D, van Berkel CG, Lemmers RJ, Frants RR, Oudejans CB, Schutgens RB, Pronk JC, van der Knaap MS. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet. 2001;29:383–8. [PubMed: 11704758]
- Leferink PS, Breeuwsma N, Bugiani M, van der Knaap MS, Heine VM. Affected astrocytes in the spinal cord of the leukodystrophy vanishing white matter. Glia. 2018;66:862–873. [PMC free article: PMC5838785] [PubMed: 29285798]
- Li W, Wang X, van der Knaap MS, Proud CG. Mutations linked to leukoencephalopathy with vanishing white matter impair the function of the eukaryotic initiation factor 2B complex in diverse ways. Mol Cell Biol. 2004;24:3295–306. [PMC free article: PMC381664] [PubMed: 15060152]
- Liu R, van der Lei HD, Wang X, Wortham NC, Tang H, van Berkel CG, Mufunde TA, Huang W, van der Knaap MS, Scheper GC, Proud CG. Severity of vanishing white matter disease does not correlate with deficits in eIF2B activity or the integrity of eIF2B complexes. Hum Mutat. 2011;32:1036–45. [PubMed: 21560189]
- Lynch DS, Rodrigues Brandão de Paiva A, Zhang WJ, Bugiardini E, Freua F, Tavares Lucato L, Macedo-Souza LI, Lakshmanan R, Kinsella JA, Merwick A, Rossor AM, Bajaj N, Herron B, McMonagle P, Morrison PJ, Hughes D, Pittman A, Laurà M, Reilly MM, Warren JD, Mummery CJ, Schott JM, Adams M, Fox NC, Murphy E, Davagnanam I, Kok F, Chataway J, Houlden H. Clinical and genetic characterization of leukoencephalopathies in adults. Brain. 2017;140:1204–11. [PMC free article: PMC5405235] [PubMed: 28334938]
- Maletkovic J, Schiffmann R, Gorospe F, Gordon ES, Mintz M, Hoffman EP, Alper G, Lynch DR, Singhal BS, Harding C, Amartino H, Brown CM, Chan A, Renaud D, Geraghty M, Jensen L, Senbil N, Kadom N, Nazarian J, Feng Y, Wang Z, Hartka T, Morizono H, Vanderver A. Genetic and clinical heterogeneity in eIF2B-related disorder. J Child Neurol. 2008;23:205–15. [PubMed: 18263758]
- Richardson JP, Mohammad SS, Pavitt GD. Mutations causing childhood ataxia with central nervous system hypomyelination reduce eukaryotic initiation factor 2B complex formation and activity. Mol Cell Biol. 2004;24:2352–63. [PMC free article: PMC355856] [PubMed: 14993275]
- Robinson MÈ, Rossignol E, Brais B, Rouleau G, Arbour JF, Bernard G. Vanishing white matter disease in French-Canadian patients from Quebec. Pediatr Neurol. 2014;51:225–32. [PubMed: 25079571]
- Scheper GC, Proud CG, van der Knaap MS. Defective translation initiation causes vanishing of cerebral white matter. Trends Mol Med. 2006;12:159–166. [PubMed: 16545608]
- Schiffmann R, Tedeschi G, Kinkel RP, Trapp BD, Frank JA, Kaneski CR, Brady RO, Barton NW, Nelson L, Yanovski JA. Leukodystrophy in patients with ovarian dysgenesis. Ann Neurol. 1997;41:654–61. [PubMed: 9153528]
- Shimada S, Miya K, Oda N, Watanabe Y, Kumada T, Sugawara M, Shimojima K, Yamamoto T. An unmasked mutation of EIF2B2 due to submicroscopic deletion of 14q24.3 in a patient with vanishing white matter disease. Am J Med Genet. 2012;158A:1771–7. [PubMed: 22678813]
- Shimada S, Shimojima K, Sangu N, Hoshino A, Hachiya Y, Ohto T, Hashi Y, Nishida K, Mitani M, Kinjo S, Tsurusaki Y, Matsumoto N, Morimoto M, Yamamoto T. Mutations in the genes encoding eukaryotic translation initiation factor 2B in Japanese patients with vanishing white matter disease. Brain Dev. 2015;37:960–6. [PubMed: 25843247]
- Song H, Haeri S, Vogel H, van der Knaap M, Van Haren K. Postmortem whole exome sequencing identifies novel EIF2B3 mutation with prenatal phenotype in 2 siblings. J Child Neurol. 2017;32:867–70. [PubMed: 28597716]
- van der Knaap MS, Bugiani M, Boor I, Proud CG, Scheper GC. Vanishing white matter. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson KM, Mitchell G, eds. Online Metabolic and Molecular Bases of Inherited Disease. Chap 235.1. New York:McGraw-Hill. 2010.
- van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol. 2006;5:413–23. [PubMed: 16632312]
- van der Knaap MS, van Berkel CG, Herms J, van Coster R, Baethmann M, Naidu S, Boltshauser E, Willemsen MA, Plecko B, Hoffmann GF, Proud CG, Scheper GC, Pronk JC. eIF2B-related disorders: antenatal onset and involvement of multiple organs. Am J Hum Genet. 2003;73:1199–207. [PMC free article: PMC1180499] [PubMed: 14566705]
- van der Knaap MS, Wevers RA, Kure S, Gabreëls FJM, Verhoeven NM, van Raaij-Selten B, Jaeken J. Increased cerebrospinal fluid glycine: a biochemical marker for a leukoencepohalopathy with vanishing white matter. J Child Neurol. 1999;14:728–31. [PubMed: 10593550]
- van der Lei HD, van Berkel CG, van Wieringen WN, Brenner C, Feigenbaum A, Mercimek-Mahmutoglu S, Philippart M, Tatli B, Wassmer E, Scheper GC, van der Knaap MS. Genotype-phenotype correlation in vanishing white matter disease. Neurology. 2010;75:1555–9. [PubMed: 20975056]
- Vanderver A, Schiffmann R, Timmons M, Kellersberger KA, Fabris D, Hoffman EP, Maletkovic J, Hathout Y. Decreased asialotransferrin in cerebrospinal fluid of patients with childhood-onset ataxia and central nervous system hypomyelination / vanishing white matter disease. Clin Chem. 2005;51:2031–42. [PubMed: 16155092]
- Wong K, Armstrong RC, Gyure KA, Morrison AL, Rodriguez D, Matalon R, Johnson AB, Wollmann R, Gilbert E, Le TQ, Bradley CA, Crutchfield K, Schiffmann R. Foamy cells with oligodendroglial phenotype in childhood ataxia with diffuse central nervous system hypomyelination syndrome. Acta Neuropathol (Berl). 2000;100:635–46. [PubMed: 11078215]
- Zhang H, Dai L, Chen N, Zang L, Leng X, Du L, Wang J, Jiang Y, Zhang F, Wu X, Wu Y. Fifteen novel EIF2B1-5 mutations identified in Chinese children with leukoencephalopathy with vanishing white matter and a long term follow-up. PLoS One. 2015;10:e0118001. [PMC free article: PMC4356545] [PubMed: 25761052]
Chapter Notes
Revision History
- 4 April 2019 (sw) Comprehensive update posted live
- 9 August 2012 (cd) Revision: multigene panel for this disorder now listed in the GeneTests™ Laboratory Directory
- 24 May 2012 (me) Comprehensive update posted live
- 9 February 2010 (me) Comprehensive update posted live
- 30 July 2007 (me) Comprehensive update posted live
- 20 February 2003 (me) Review posted live
- 19 November 2002 (pb) Original submission
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: February 20, 2003; Last Update: April 4, 2019.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
van der Knaap MS, Fogli A, Boespflug-Tanguy O, et al. Childhood Ataxia with Central Nervous System Hypomyelination / Vanishing White Matter. 2003 Feb 20 [Updated 2019 Apr 4]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

