© 2023 John Mattick and Paulo Amaral.
Open Access: This content is Open Access under the Creative Commons license CC-BY-NC-ND.
Bookshelf ID: NBK595928DOI: 10.1201/9781003109242-7

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mattick J, Amaral P. RNA, the Epicenter of Genetic Information: A new understanding of molecular biology. Abingdon (UK): CRC Press; 2022 Sep 20. doi: 10.1201/9781003109242-7

By the 1970s, it was evident that most sequences in the genomes of complex organisms are not protein-coding. The amount of cellular DNA was found to broadly increase with developmental complexity, but there were incongruities, termed the C-value paradox. Theoretical considerations of the lethality of protein-coding mutations, the presence of large amounts of repetitive sequences and seemingly defective ‘pseudogenes’ all suggested that some, and perhaps most, multicellular organisms carry variable amounts of superfluous ‘non-informational’ DNA. The corollary of neutral evolution of non-functional sequences was widely accepted, although there was debate between the ‘near-neutralists’ and ‘adaptionists’ concerning the signatures of protein-coding genes and regulatory sequences underpinning quantitative trait variation. Later analyses showed many different rate classes of sequence evolution in plant and animal genomes. Nonetheless, there was growing consensus that much if not most of the DNA in plant and animal genomes must be junk and that the many repetitive sequences, by then known to be derived from transposons, are ‘selfish’ genetic hobos and freeloading passengers. The discovery in 1977 that eukaryotic genes are composed of short fragments of protein-coding sequences (‘exons’) interspersed with non-coding sequences (‘introns’), which are removed by post-transcriptional splicing, explained heterogeneous nuclear RNA and was proffered as further evidence of junk. Introns were rationalized as extant remnants of the prebiotic assembly of genes, which had been deleted from microbial genomes under selective pressure for rapid replication, but persisted and were fattened by transposons in complex organisms, despite the fact that complex organisms have microbial ancestors. Introns were found to increase in number and size with developmentally complexity, which suggested that they may have evolved developmentally important functions transacted by RNA.
Following Avery’s demonstration that DNA is the genetic material, cytological and biochemical measurements of the amount of cellular DNA showed that species have a characteristic DNA content a (termed the ‘C-value’ by Hewson Swift 1 ) and that the amount of cellular DNA broadly increases with developmental complexity if taxa are compared on the basis of their minimal DNA content. 2 , 3 Related studies during this period used the drug colchicine to block DNA replication at metaphase, enabling the complement and size distribution of chromosomes also to be determined. b
However, anomalies were found. In many taxa, the cellular DNA content of species varies over a wide range: 6 , 7 some simple protists and plants such as green algae and mosses have more DNA than flowering plants; and many plants (including onions, a popular example 8 ) and some other protozoans such as amoebae have more DNA per cell than mammals 3 , 9 , 10 (Figure 7.1). Since it was assumed that more complex organisms c require more genetic information (and the understanding of gene structure and regulation was derived from microbial genetics and biochemistry studies), these anomalies led to the coining of the term ‘C-value paradox’ 9 or ‘C-value enigma’. 7

The range of haploid genome sizes for the groups of organisms listed. (Adapted from an image by Steven Carr, Memorial University of Newfoundland, published in Fedoroff, with permission from the American Association for the Advancement of Science.)
There has been, and remains, considerable speculation about the significance of the spectrum of DNA content, which is often interpreted as evidence of the ability of eukaryotes to maintain superfluous DNA. 7 Correlations were sought and sometimes found with cell size, involving an increase in the number of nuclei and/or copies of the genome, possibly to support the metabolism of larger cells. 11–15
The inordinately large amounts of DNA in some species transpired to be due to two factors: polyploidy, i.e., multiple copies of the genome, which occurs commonly in plants and sporadically in animals, especially insects; 17–21 and lineage-specific expansions of transposon-derived sequences, notably in some fishes and amphibians (especially lungfish 22 and salamanders 23 ), d some clades of arthropods 24 , 25 and cnidarians (hydra), 26 and many plants, where they play a major role in adaptive evolution (Chapter 10). e
The G-value enigma emerged later, when the genome projects showed that there is no correlation between the number of protein-coding genes and developmental complexity (Chapter 10). Genome sequencing also showed that the ratio of non-protein-coding to protein-coding DNA (which intrinsically corrects for ploidy) increases with morphological complexity, 21 , 27 suggesting that, whatever else may be at play, increased complexity is associated with the expansion of regulatory information. This imputation can only be falsified by a downward exception, i.e., the identification of developmentally complex organisms that have little non-coding DNA, of which none have been found to date. f
For decades, however, the notion that “the number of distinct protein-coding genes that an organism made use of was a valid measure of its complexity” was deeply rooted and well accepted. 28
The mechanisms by which genomes can be enlarged are gene, segmental or whole genome duplication – first proposed by Susumo Ohno in 1970 29 – and copy-and-paste insertion of sequences from external sources or elsewhere in the genome by transposition. That is, the raw material for evolutionary innovation is sequence duplication and transposition. The former has been documented in many species, for example, in yeast and at the origin of the vertebrates, where it is evident that whole genome duplication has occurred at some point in their evolutionary history, with some duplicated genes having acquired new functions and been retained, whereas those that remained redundant were largely lost. 30–32 Partial genome (segmental) duplication is also well documented. 33
The work of Leslie Gottlieb, Donald Levin and others has shown that genome duplication (‘autopolyploidy’) and fusion of genomes between related species (termed ‘allopolyploidy’) g creates phenotypic novelty and speciation – altering patterns of gene expression, physiological responses, growth rates, developmental features, reproductive outputs, mating systems and ecological tolerances, 34–36 including in Darwin’s finches. 37 This led Levin to suggest that such nucleotypic effects may “‘propel’ a population into a new adaptive sphere, perhaps accounting for the distribution of polyploids, both auto- and allopolyploids, in areas beyond those of the diploid parents”. 36
Transposition is a specialized and highly flexible form of sequence relocation or (more commonly) ‘duplication’ (multiplication) that mobilizes protein-coding and/or regulatory cassettes, 38–42 which explains its evolutionary value and distribution (see below). Transposases are, in fact, the most abundant and ubiquitous genes in nature. 43 Large numbers of various classes of retrotransposed sequences occur in multicellular eukaryotes, especially plants and animals 44 , 45 and the colonization of genomes by transposons appears to have occurred in bursts 45 likely associated with major evolutionary adaptations (see, e.g., 22 , 46 ) (Chapter 10). Transposable elements (TEs) are diverse and have been widely incorporated into regulatory networks in different clades, 47 as predicted by Britten and Davidson (Chapter 5), with, for example, most primate-specific regulatory sequences having been derived from these elements. 38 , 39
It is reasonable to assume that genomes contain some duplicated or transposed sequences that are in suspension between functional exaptation on the one hand and degradation or deletion on the other, i.e., have not (yet) acquired a useful (new) function nor been lost. It is currently difficult, if not impossible, to determine the extent of such limbo sequences in any given lineage. One might speculate, however, that the more ancient the duplication or TE, the more likely it is to have acquired, or already have, a useful function that has contributed to its retention. One might also speculate that recently acquired transposable elements have played a role on phenotypic diversification, which has now been well documented 39 , 48 (Chapter 10).
The problem was that the large genomes of protists, plants and animals, and their large numbers of ‘repetitive’ sequences, could not be reconciled with the protein-centric conception of genetic information.
The population geneticists and evolutionary theorists at the time, notably Müller h and Ohno, suggested that, since increases in genome sizes in eukaryotes occurred by polyploidization, much of the duplicated DNA is redundant. They also argued that, if the unique sequences (~50% of the genome) in mammals specified structural (i.e., protein-coding) genes, there would be ~1 million such genes, which would, by comparison with bacteria, impose an unbearable mutational load, the escape from which was the prime function of recombination. 49 , 51–53
Ohno extended this logic to regulatory information, speculating that “in order not to be burdened with an unbearable mutation load, the necessary increase in the number of regulatory systems had to be compensated by simplification of each regulatory system. It would not be surprising if each mammalian regulatory system is shown to have fewer components than the lac-operon system of Escherichia coli”. 53
Based on these considerations and Haldane’s 1957 ‘cost of selection’ principle, which stated that the number of gene loci in a genome is a key determinant of the rate of evolution, 54 Masatoshi Nei concluded in 1969 that, given the “high probability of accumulating … lethal mutations in duplicated genomes... it is to be expected that higher organisms carry a considerable number of nonfunctional genes (nonsense DNA) in their genome” and that “higher organisms, including man … are using only a small fraction of the maximum amount of genetic information their DNA molecules are able to store”. 55 , 56 This logic has persisted to the present, 8 , 57 underpinning the recent claim, for example, that “the functional fraction within the human genome cannot exceed 15%”. 58
Such early musings were based on the analysis of easily discernible simple traits, which constituted the majority of genetic studies up until that time and indeed until the end of the 20th century. These traits included metabolic defects, i flower and eye color j and severe genetic disorders, which usually result from high-impact loss-of-function mutations in protein-coding sequences. By and large, they did not take into account that variations in regulatory sequences that control quantitative traits in complex organisms may be more subtle, although they may have a strong influence on complex traits and reproductive fitness: this was a huge blind spot.
In this context, it should be noted that the mathematical foundations of quantitative genetics were laid down with a very different set of problems in mind – such as the prediction of short-term responses to artificial selection – which went on to focus on genetic diversity based on enzyme polymorphisms, 59 again before crucial details of the variation in genome sequences and of genome regulation in complex organisms were known. 60
Incorporating molecular considerations, John Paul (1972) stated the alternatives that, considering the existence of hnRNAs and the size of mammalian genomes, “either that the mutational load argument does not hold for eukaryotes or [as concluded by others] that much of the DNA in eukaryotes is not informational”. 61
He speculated that the more and less compact regions of chromosomes differ chemically, in that “modified histones, modified DNA or extra substances” determined the conformation of ‘nucleohistone’ (chromatin). He reasoned that non-histone proteins would perform this function, with auxiliary participation of nascent RNAs. In his model, “address sites” in the interbands would be targets for “polyanionic” regulators, allowing relaxation and transcription of nascent RNA that would not only contain an mRNA sequence but would also accumulate in these regions, recruiting RNA binding proteins and inducing further unwinding of chromatin.
Paul also used this possible role for nascent transcripts to explain the existence of the very large transcriptional units (hnRNAs) in animals and plants, which would contain the sequence of mRNAs together with redundant sequences producing ‘nonsense RNAs’ that perform an ‘unwinding role’. Although vaguely defined, this was one of the first models that posited RNAs and histone modifications acting together to regulate gene expression. This model also predicted, as did others (Chapter 5), that these nascent RNAs are processed to generate mRNAs, and even suggested the existence of sequence signals in the hnRNAs that guide the processing into the RNA parts to be degraded in the nucleus or to be exported to the cytoplasm. 61
The issue was summarized by Ed Southern in 1974: “The outstanding problem presented by eukaryotic DNA is that of finding a role for these large fractions not used in coding for proteins or cytoplasmic RNAs.” 62
Speculation was rife. The evolutionary biologist Tom Cavalier-Smith wrote in 1978:
Eukaryote DNA can be divided into genic DNA (G-DNA), which codes for proteins (or serves as recognition sites for proteins involved in transcription, replication and recombination), and nucleoskeletal DNA (S-DNA) which exists only because of its nucleoskeletal role in determining the nuclear volume … 11 , 12
Others suggested the excess non-coding DNA might be retained for “genome balance”, 63 have some value as a mutational sponge 49 or buffering, 64 or be a reservoir for evolutionary innovation. 65–68
A natural corollary of the idea that much of the genomes of plants and animals is not functional is that these sequences are evolving ‘neutrally’. In parallel, the growing availability of amino acid sequence data revealed that protein sequences have been diverging between lineages at a relatively constant rate, k referred to as the “molecular clock” by Emile Zuckerkandl and Linus Pauling, 71 or “genetic equidistance” by Emanuel Margoliash 72 in the early 1960s. In 1971, Richard Dickerson showed that the clock runs at different rates for different proteins 73 (Figure 7.2), later shown to vary by orders of magnitude, useful to measure evolutionary relationships over different genetic distances and evolutionary timescales. 74–76

The molecular clock. Dickerson's graph of the rates of molecular evolution in fibrinopeptides, hemoglobin and cytochrome c. (Reproduced from Dickerson with permission from Springer Nature.)
The divergence of the sequences of homologous proteins over time was surprising and, after taking into account the frequencies of deleterious mutations and mutational load, led Motoo Kimura to propose in 1968 the neutral theory of molecular evolution, or ‘genetic drift’, which posited that “an appreciable fraction” of the genome was evolving independently of natural selection. 77–81 Like Nei, Kimura was also motivated by Haldane’s argument and the 1970s finding that the numbers of nucleotide changes observed between humans and chimpanzees could not be explained by selection, called “Haldane’s Dilemma”, 82 tacitly assuming that mutation is random and not influenced by other mechanisms (see Chapter 18).
A similar proposal was made by Jack King and Thomas Jukes in their 1969 article entitled ‘Non-Darwinian Evolution,’ which extolled the importance of random genetic changes and genetic drift in evolution. 50 The theory was refined in 1973 by Kimura’s student, Tomoko Ohta, and later by others, notably Michael Lynch, who emphasized the importance of nearly neutral (“slightly deleterious”) mutations, whose exposure to selection is dependent on the size of the interbreeding population. 83–88
The extension of this logic, posited as the ‘null’ hypothesis, 89 is that highly complex organisms with small effective population sizes such as mammals accumulate greater loads of transposable elements, larger introns (see below) and larger intergenic regions, all of which co-vary inversely with population size, such that especially large bodied species with low population sizes have bloated genomes and difficulty in purging even slightly deleterious mutations. 85–88 , 90 Later theoretical studies also concluded, mainly based on ‘non-conservation’, that alternative transcription, polyadenylation, RNA modification and RNA editing l sites in complex organisms are non-adaptive. 91–95
Neutral evolution was controversial in evolutionary circles, reflecting a long-standing disagreement between ‘classical’ and ‘quantitative’ geneticists that simmered for decades, although thought to have been resolved by Fisher’s infinitesimal model 96 (Chapter 2). The classical geneticists viewed the normal state to be a wildtype (protein-coding) gene with a low frequency of deleterious (usually recessive) mutants in the population, influenced by Mendel’s simple trait segregation in peas and by genetic (‘Mendelian’) disorders in humans. On the other hand, the quantitative geneticists, mainly working in agriculture, citing the abundant variation in quantitative traits in crop plants and livestock and the ‘concealed variability’ revealed by inbreeding experiments, proposed that many genes have two or more alleles maintained at intermediate frequencies in populations by ‘balancing’ selection, perhaps influenced by environmental factors. 59
A renewed debate between the ‘near-neutralists’ and ‘adaptationists’ ensued following Kimura’s and Ohta’s papers. 97 The former maintained that genetic drift accounts for most differences within populations or between species, whereas the latter credited them to positive selection for adaptive traits, 98 , 99 although as Laurence Hurst later observed “the two positions are often hard to discriminate as they make many similar predictions”. 97
These debates did not often consider that there might be an important distinction between the genetic signatures of protein and regulatory variation and were mostly thought of in terms of binary (wildtype and ‘defective’) alleles rather than interconnected networks. 99 , 100 Nor did they take into account the role of transposons in phenotypic variation (see below; Chapters 5 and 10), positive selection for reproductive success, 99 , 100 or the amount of information that might be required to organize the four-dimensional development of multicellular organisms 101 (Chapter 15). Moreover, nearly neutral genetic drift does not account for the rapid evolution of animal phyla and species, such as observed in the Cambrian explosion, Darwin’s finches 102 and primates, 103–109 and is at odds with the whole genome biochemical indices of function that were revealed later (Chapter 13). m
As Mayr observed in 1970:
The day will come when much of population genetics will have to be rewritten in terms of interaction between regulator and structural genes. This will be one more nail in the coffin of beanbag genetics. It will lead to a strong reinforcement of the concept that the genotype of the individual is a whole and that the genes of a gene pool form a unit. 110
And Jacob in 1977:
It seems likely that divergence and specialization of mammals, for instance, resulted from mutations altering regulatory circuits rather than chemical structures. Small changes modifying the distribution in time and space of the same structures are sufficient to affect deeply the form, the functioning, and the behavior of the final product – the adult animal. 111
The situation was summarized in 2014 by Karl Niklas:
Beginning with a series of papers in the early 20th century and culminating with his book The Genetical Theory of Natural Selection, Ronald A. Fisher (1930) founded the field of population genetics and designated the gene as the unit of stable hereditary transmission between successive generations. This genocentric view of inheritance asserted the preeminent importance of allele frequency distributions and differential reproductive success in evolutionary processes. However, it failed to explore alternative origins of phenotypic variation. It simply assumed that all phenotypic variants result from [protein-coding] gene mutations … Perhaps even more restrictive was the additional assumption that the phenotype could be mapped directly onto the genotype and thus described simply by changes exclusively at the level of individual genes or sets of genes. 112
Niklas continued:
This outlook was challenged in the 1970s and 1980s within a field of study soon to be called evolutionary‐developmental biology, or simply evo‐devo, which asserted that evolutionary phenotypic transformations are the result of changes in gene expression patterns rather than the immediate products of mutations of individual genes … Arguably … this perspective can be traced back to a seminal paper by Britten and Davidson. 112
Richard Lewontin, who developed some of the statistical tools for assessing genetic drift and selection (largely from studies of electrophoretic variation in proteins in natural populations of Drosophila), 59 observed in 1974 that
For many years population genetics was an immensely rich and powerful theory with virtually no suitable facts on which to operate. It was like a complex and exquisite machine, designed to process a raw material that no one had succeeded in mining… Quite suddenly the situation has changed. The mother-lode has been tapped and facts in profusion have been poured into the hoppers of this theory machine. And from the other end has issued – nothing … The entire relationship between the theory and the facts needs to be reconsidered. 98
In 1996, Ohta admitted, with respect to nucleotide substitution patterns, that “all current theoretical models suffer either from assumptions that are not quite realistic or from an inability to account readily for all phenomena”. 113
The concept of neutral evolution led to attempts to define a subset of sequences that are evolving neutrally, to measure the unconstrained rate of sequence drift, and thereby determine which (other) sequences in the genome might be evolving more rapidly or slowly under positive or negative selection, n and therefore be functional.
One obvious candidate was the ‘redundant’ (usually third) base of synonymous codons, first exposed by the pioneering sequencing in 1983 of 11 cloned alcohol dehydrogenase (Adh) genes in natural populations of Drosophila, which revealed 43 previously hidden polymorphisms. Only one of these polymorphisms altered a codon specificity (and resulted in the known electrophoretic variant of the protein), implying that nonsynonymous changes have phenotypic consequences and are deleterious, whereas the others were possibly neutral. 114 However, later analyses showed that amino acid codon sequences are not evolving neutrally (as is also the case for many non-coding sequences), possibly reflecting selection pressures on translational efficiency or RNA structure, 115–119 with others showing that the genetic code is optimal for encoding additional information. 120–122 Later studies showed that non-coding polymorphisms affect Adh expression 123 and that Adh variants are selected indirectly. 124
The field also looked to other sequences, notably ‘pseudogenes’ (see below) and ancient retrotransposons, to estimate the rate of neutral evolution, on the questionable and likely incorrect assumption that they are non-functional (Chapter 10), leading to a vast underestimation of the amount of the human genome that is under selection (Chapter 11).
The debate continues, 125 , 126 but the concept of neutral evolution is coming under siege. As concluded recently by Andrew Kern and Matthew Hahn:
The neutral theory was supported by unreliable theoretical and empirical evidence from the beginning, and … we argue that, with modern data in hand, each of the original lines of evidence for the neutral theory are now falsified, and that genomes are shaped in prominent ways by the direct and indirect consequences of natural selection. 127
The adherents begged to differ. 128
Of course, different types of sequences have different structure-function constraints and different selection pressures, as is seen within protein-coding sequences where the amino acid sequences of active sites are highly conserved but associated scaffolding and domain linker sequences are quite plastic. 129 , 130 Regulatory sequences are even more plastic; 131–135 orthologous promoters that have no obvious sequence homology direct similar expression patterns in fish and humans, 136 and less than 5% of human embryonic stem cell developmental ‘enhancers’ (Chapter 14) are ‘conserved’ in mouse. 135 These regulatory sequences also encompass small regulatory RNAs and vast numbers of tissue- and cell-type specific long non-coding RNAs, which seem to be even more evolutionarily flexible, with different sequence-structure-function constraints and including increasing numbers of functionally validated species- and clade-restricted RNAs (Chapters 12, 13 and 16).
It is now well established that adaptive radiation in complex organisms, including primates, is mostly due to regulatory variation, 137–142 which may be co-dominant and therefore immediately visible to selection. 143 Regulatory sequences evolve rapidly, o mostly (initially at least) under positive selection for changes in morphological and physiological phenotypes.
It has also been known for some time, confirmed later by the genome projects (Chapters 10 and 11), that the mutation spectrum varies enormously across the genome, 144 , 145 which has been rationalized for example as local variation in the underlying mutation rate (due to regional differences in nucleotide composition) or the activity of DNA repair enzymes. 144–147 The alternative explanation that the vast non-coding regions of plant and animal, especially mammalian, genomes are under selection could not be countenanced, both because it was assumed to be junk and because it appeared impossible due to the mathematical models of selection operating on random mutation in small populations.
A later analysis showed that there are at least seven different rate classes of sequence evolution in the mammalian genome, 148 and different rates of sequence evolution in gene promoters. 149 Others concluded that ~95% of the human genome is influenced by background selection and biased gene conversion, 150 commensurate with the proportion of the genome that is dynamically transcribed into (mainly non-protein-coding) RNA (Chapter 13), while observations in natural Arabidopsis accessions show that epigenome-associated mutation bias occurs differentially across the genome and gene regions, with essential genes (in particular gene bodies) subject to stronger purifying selection having a lower mutation rate. 151 , 152
It did not seem to occur to most at the time, apart from McClintock, Britten and Davidson and a few others (Chapter 5), that the enormous numbers of ‘repetitive’ sequences and nuclear-localized RNAs might play a role in plant and animal differentiation and development.
And since no one could countenance gigabases of regulatory protein-binding sites, p and for all of the other reasons cited above, Ohno summed up the growing consensus when in 1972 he wrote about “all that ‘junk’ DNA in our genome” q arguing that only a fraction of the human DNA functions as ‘genes’ and that there is “more than 90% degeneracy contained within our genome”. 49 Ohno’s conclusions were reinforced by the existence of seemingly defective ‘pseudogenes’ 161 (first identified in 1977 and described as ‘relics of evolution’ 162 ), the ‘gene-poor’ and transposable element-rich heterochromatin, and the extensive intergenic regions in intensively studied loci, thought to be genetically and transcriptionally silent but later shown to be the sites of ‘enhancer’ and other regulatory elements that control gene expression patterns in development (Chapters 14 and 16).
Duplications of globin genes were highlighted by Ohno and others not only as the source of new functional genes, but also of defective pseudogenes with untranslatable sequences (“recent degenerates”), 49 , 163 many of which have since been shown to have regulatory functions 164 (Chapters 10 and 13). r Notably, the pseudogene in the human hemoglobin cluster on chromosome 11, hemoglobin subunit beta pseudogene 1 (HBBP1, or η-globin pseudogene in primates), was later found to be subject to strong selection, tissue-specifically expressed, essential for erythropoiesis, mutated in a form of thalassemia and to regulate the switches of globin gene expression during development. 170–173
Some did take issue. Herb Boyer questioned the calculations of the extent of functionality in the genome based on the assumption that lethal mutation rates apply to the whole genome, noting in the discussion of Ohno’s paper that “we can only measure what we see”. 49
Stephen O’Brien wrote shortly afterwards that the
conclusions (that) indicate that more than 90% of the eukaryotic genome may be composed of nonfunctional or noninformational ‘junk’ DNA … have not been fundamentally proven; rather they are based on simplifying assumptions of questionable validity, in some cases contradictory to experimental data. 174
He challenged the notion that lethal mutation frequency was a good metric for gene number and genome functionality, citing several lines of evidence suggesting that mutations only result in lethality in a minority of genes. s Noting that hybridization studies indicate the presence of a minimum of 300,000 different transcripts of 1 kb or more in mouse brain alone, he made the very reasonable point that “RNA does not have to be translated to have a function” and that their existence and their tissue- and developmental stage-specific expression transcription support their functionality. 174
Heterochromatin was also widely thought to be inert, despite the fact that it is dynamic, and there was, even then, considerable evidence of its importance in developmental processes. 177 As observed by Spencer Brown in his incisive 1966 paper: “Our present picture of gene action comes almost exclusively from microorganisms. It is a verbally simply one … the systems controlling gene regulation in higher organisms probably involve highly complex mechanisms necessary for developmental integration.” Among several considerations on the potential roles of heterochromatin, he noted the reports of chromatin associated RNAs and pondered regarding the abundant RNAs present exclusively in the nucleus that “Such observations would make sense if the genes in higher organisms were required to build complex machinery for their own control”. 177
Jim Peacock and colleagues pointed out in 1978:
In recent years it has become clear that specific genetic properties are attributable to heterochromatic regions of chromosomes and that the different segments of heterochromatin in a genome may have different properties … we present data, primarily from Drosophila, to show that the heterochromatin of each chromosome has a unique, segmental identity, and that DNA sequences in heterochromatin have, as do DNA sequences in euchromatin, defined patterns of conservation and change during evolution. We show that the properties discovered in Drosophila apply to other eukaryotes, including plants and mammals. 178
Put simply, the use of the frequency of lethal mutations by evolutionary theorists, the emphasis on negative selection to assess the extent of functionality of the genomes of complex organisms, and the assumptions that repetitive sequences and pseudogenes are non-functional were based on only rudimentary knowledge of molecular genetic information and were biased by emphasizing deleterious mutations over quantitative trait variations. It was conceptually primitive, but unfortunately influential.
Cloning and more advanced genetic mapping techniques t would later show the majority of mutations that cause severe phenotypic consequences in mammals map to protein-coding sequences – what might be called ‘catastrophic component damage’. However, the vast majority (~95%) of variations affecting complex traits – with few or only subtle effects on viability – occur outside of protein-coding sequences (Chapter 11). This is largely invisible to high-fitness-impact and lethality-based measures of genetic load but constitutes the more important component of phenotypic variability in natural populations.
However, Mayr, O’Brien and others were swimming against the tide. The phrase ‘junk DNA’ entered the popular lexicon, uncritically embraced by those – including Brenner 65 – who were convinced of the primacy of proteins in the specification of cell and developmental biology, seemingly incurious about what all that non-protein-coding DNA might be doing. In fact, as seen below, proponents of junk DNA explicitly discouraged research into the possible roles of non-coding regions of genomes.
A logical extension of the junk DNA view was the proposal promulgated and popularized by Richard Dawkins in 1976, following earlier theorizing by George Williams 179 and William Hamilton, 180 that DNA sequences have a propensity to select for their survival, which he termed “the selfish gene”. 181 Dawkins argued that the selfish gene hypothesis can explain the fact that “a large fraction of the DNA is never translated into protein”, stating “The simplest way to explain the surplus DNA is to suppose that it is a parasite, or at best a harmless but useless passenger, hitching a ride in the survival machines created by the other DNA”. 181
The concept was extended in 1980 by back-to-back papers by Ford Doolittle and Carmen Sapienza, and Leslie Orgel and Crick, entitled ‘Selfish genes, the phenotype paradigm and genome evolution’ and ‘Selfish DNA: the ultimate parasite’, respectively. 155 , 182 , 183 As put by the latter:
In summary, then, there is a large amount of evidence which suggests, but does not prove, that much DNA in higher organisms is little better than junk. We shall assume, for the rest of this article, that this hypothesis is true … What we would stress is that not all selfish DNA is likely to become useful. Much of it may have no specific function at all. It would be folly in such cases to hunt obsessively for one. 183
The exemplars of selfish DNA were sequences derived from (endogenous) retroviruses, transposons and other types of repetitive elements (Figure 7.3), which reinforced the view that these elements are (mainly) genetic hobos, u notwithstanding McClintock’s demonstrations that transposon mobilization changes developmental phenotype and responses to the environment. 184 The view of transposons as functionless and/or deleterious parasites v has endured, 189–191 reinforced by the discovery that they are restrained by methylation (Chapter 14) and other ‘silencing’ mechanisms. Although their discovery earned McClintock a Nobel Prize in 1983, it seems that her emphasis and insistence on them as controlling elements in differentiation and development, and the finding that TEs cause insertional mutations in bacteria, delayed the award. 192 , 193

Classification of transposable elements and mechanisms of transposition. Class I retrotransposons mobilize via an RNA intermediate. Class II DNA transposons utilize a DNA intermediate. Autonomous elements encode the enzymatic machinery necessary for their (more...)
The role of transposons as mobile cassettes of genetic (especially regulatory) information in evolutionary and biological processes, and the proportion of transposon-derived sequences that may have contributed or acquired useful functions in genomes 194 was then and is still not known, but it suited the zeitgeist to assume that it is low. The selfish fattening out by transposons of intergenic sequences and introns within genes (see below) then became the widely accepted explanation for all that junk, and the C-value enigma. 182 , 195 , 196
Perhaps the most unexpected discovery in the history of molecular biology was that genes in eukaryotes, especially developmentally complex eukaryotes, are not co-linear with their encoded proteins, but rather are fragmented and separated by non-protein-coding ‘intervening’ sequences or ‘intragenic regions’, dubbed ‘introns’. 157 , 197 , 198 The fragments of protein-coding sequences (and flanking regulatory sequences in mRNAs) were reciprocally called ‘exons’. w
In 1975, Darnell and colleagues showed that adenovirus mRNA is derived from a high molecular weight precursor. 199 In 1977, Phillip Sharp and Rich Roberts and their colleagues observed under the electron microscope that adenovirus mRNAs do not hybridize contiguously to the adenovirus genome, but rather loop out in segments (Figure 7.4), indicating that the mRNA is derived from regions of the genome that are not adjacent, 200–202 confirmed by others. 203
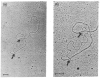
Electron micrographs of a hybrid between and adenovirus-derived mRNA and adenovirus DNA, with arrows showing boundaries of the R-loop of single-stranded DNA that is not present in the mRNA, the first demonstration of the presence of introns. (Reproduced (more...)
The same phenomenon was soon reported in vertebrate genes encoding β-globin, 204 , 205 chicken ovalbumin (Figure 7.5) and lysozyme, 206–208 and immunoglobulin light chain, 209 and in ribosomal RNA genes in Drosophila, 210 whose cloned mRNA (cDNA or complementary DNA) sequences hybridized to multiple larger sized fragments of restriction endonuclease-digested genomic DNA in Southern blots. This is impossible to explain unless the cDNA sequences were spread over a large section of genomic real estate, which was confirmed by transcript mapping and sequence analysis. 211–213
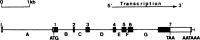
Exon intron structure of the chicken ovalbumin Y gene. Filled boxes indicate protein-coding sequences, with unfilled areas indicating 5′ and 3′ untranslated regions in the mature mRNA. (Reproduced from Heilig et al. with permission from (more...)
It transpired that the intervening sequences are ‘spliced’ out from the primary transcripts 197 , 202 , 214 , 215 – now called pre-mRNAs – in the nucleus, by a complex RNA-guided and catalyzed process (see following chapter), which explained the previously observed hnRNAs. The re-assembled ‘mature’ mRNAs are then exported to the cytoplasm for translation into proteins, so all was right with the gene = protein worldview, even if it is stranger than could possibly have been imagined.
The discovery of split genes (or “genes-in-pieces” as phrased by Walter Gilbert 157 ) and mRNA splicing in eukaryotic cells was “a complete shock to the scientific world”, as it broke another fundamental tenet of gene expression – the concept of collinearity – as “everyone assumed that the structure of a gene was a contiguous string of base pairs, from which information was transferred for synthesis of a protein”. 215
The presence of introns interrupting the mRNA sequences of eukaryotic genes was immediately and universally assumed to be another manifestation of, and proffered as further evidence for, junk DNA 155 , 182 , 183 , 216 – notwithstanding and not considering the obvious alternative that other information may be transmitted by the excised non-protein-coding RNA, 217 and contemporary reports of “intron-mediated enhancement” of gene expression. 218 That the possibility that introns or intronic RNAs contain functional signals was not canvassed at the time is testimony to the strength of the belief that genetic information is (only) transduced through proteins, entrenched just 16 years after the lac operon.
Nonetheless the discovery of introns meant that the mystery of mammalian mRNA biogenesis had been solved. 219 It also helped to explain the vast amount of non-coding DNA in the genomes of higher organisms, and the existence of hnRNAs and the excess of RNA in the nucleus, reconciling the Central Dogma with these unusual features of eukaryotic gene expression. Crick described introns as “‘nonsense’ stretches of DNA interspersed within the sense DNA”. 214 As put by Ohno, while bacterial genomes are “small and tidy”, filled with polycistronic genes, the genomes of vertebrates are “untidy to the extreme”, with genes spaced very apart from each other in such a way that “translation through so long spacer is out of question … [and] there was no choice but to achieve the fusion of adjacent coding sequences at the post-transcriptional level”. 220
It was also assumed that the excised intronic RNA is quickly degraded and the ribonucleotides recycled, although the technology of the time was too primitive to draw this conclusion. Northern blots, which may have been able to track the fate of the excised RNAs, had just been introduced (Chapter 7) and often relied on polyA-based purification protocols that neither capture nor detect spliced out RNAs. x Sharp and colleagues stated (with supporting references) that “introns are excised from pre-mRNAs with a half-life of 3 seconds to 30 seconds” 224 but a year later asserted (without supporting references) that excised introns “are rapidly degraded… (with) a half-life of … the order of a few seconds”, 225 an entirely different statement, indicative of the logical slip. In fact, intron-specific in situ hybridization showed that excised intronic RNAs can be relatively stable and easily detectable in the nucleus. 226 Later studies showed that many functional RNAs are derived from introns (Chapter 8) and that intronic RNAs – including retained introns and intron-derived RNAs – constitute the major fraction of the non-coding RNAs in mammalian cells. 227
Sweeping introns under the intellectual carpet as junk (like the transposon-derived sequences within them) still left the question of how the split gene arrangement came to be in the first place. Their existence was subsequently rationalized by Gilbert as a hangover of the primordial assembly of genes from fragments of protein-coding information (the ‘introns-early hypothesis’). 157
Gilbert also predicted that the presence of introns would enable ‘alternative’ splicing and thereby the evolution of modularity in protein-coding genes, expanding the repertoire of protein isoforms in complex organisms 157 (Figure 7.6). This proved to be correct, 229–232 and was recently shown to include the exonic capture of fragments of transposable elements to allow the protein to act as a genome-wide transcriptional regulator, leading to the conclusion that “TEs interacting within their host genome provide the raw material to generate new combinations of functional domains that can be selected upon and incorporated within the hierarchical cellular network”. 41
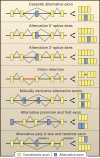
Types of alternative splicing. (Reproduced from Blencowe with permission of Elsevier.)
Gilbert’s hypothesis was elaborated independently by Darnell, 233 Doolittle 234 and Colin Blake, 235 with the sequitur that exons would be predicted to encode protein functional units or “smaller, supersecondary structures”. 235 While there was evidence that some exons corresponded to protein domains, 236–238 it was difficult to show that most protein-coding exons comprised modular elements of protein structure, 217 , 239 and later studies showed that alternative splicing is more common in regulatory sequences in mRNAs and non-coding RNAs than in protein-coding exons. 240
Developmentally complex organisms have a greater number and larger size of introns, 241 , 242 comprising at least 40% of the human genome – and likely much more, given that there are many distal alternative promoters and 5′ exons expressed in early development, introns in genes encoding non-coding RNAs, and many genes enclosed within introns of other genes (Chapter 13).
By contrast, it was argued, the genomes of fast-growing microorganisms had been streamlined under pressure for rapid replication, overlooking the fact that developmentally complex eukaryotes had microbial ancestors for at least a billion years, which would have been subject to the same pressures. As Gilbert expressed it: “… introns were lost in the course of evolution … [and] only genes in slowly replicating cells of complex organisms still retain the full stigmata of their birth”. 243 This is, in evolutionary terms, a non sequitur, but nonetheless was repeated by others. Brenner in 1990:
There is a view that E. coli is primitive and we are advanced. That is true from the point of view of function and action. But it is not true from the point of view of genome structure. Here it is E. coli that is streamlined and sophisticated, whereas it is our genome that has preserved a far more primitive condition. 244
Introns were later found to reside in out-of-the-way places (non-translated RNA genes) in bacteria, 245 to have self-splicing capability and to be able to act as mobile genetic elements 246 (Chapter 8). Cavalier-Smith, Norman Palmer and John Logsdon Jr. argued that it was more likely that introns entered (by reverse splicing, Chapter 8) and expanded in complex organisms late in evolution, 216 , 241 , 247 , 248 while not challenging the assumption that they are devoid of information. This view persisted despite the examples of conserved sequences within them, which if removed or mutated have phenotypic effects (and, as seen later, also encoding distinct and stable RNA species). 249–252
The prevailing view was summarized by Matt Ridley in his 1999 book Genome: The Autobiography of a Species in 23 Chapters:
Each gene is far more complicated than it needs to be, it is broken up into many different ‘paragraphs’ (called exons) and in between lie long stretches (called introns) of random nonsense and repetitive bursts of wholly irrelevant sense, some of which contain real genes of a completely different (and sinister) kind … But ninety-seven per cent of our genome does not consist of true genes at all. It consists of a menagerie of strange entities called pseudogenes, retropseudogenes, satellites, minisatellites, microsatellites, transposons and retrotransposons: all collectively known as ‘junk DNA’, or sometimes, probably more accurately, as ‘selfish DNA’. Some of these are genes of a special kind, but most are just chunks of DNA that are never transcribed into the language of protein. 253
The presumed irrelevance of the vast tracts of transcribed non-protein-coding RNAs – “Mother Nature’s dirty little secret” 253 or “junk in the attic” 65 – became accepted as such.
There was an alternative, equally if not more plausible, and far more interesting, possibility canvassed by John Mattick in 1994, 217 i.e., that the separation of transcription from translation in eukaryotes allowed the invasion of protein-coding genes y by introns. 246 , 254 He posited that the evolution of the spliceosome then allowed these sequences to explore new genetic space and to acquire functions as RNA regulatory signals (or ‘informational RNA’, iRNA) expressed in parallel, akin to efference signals in neurobiology, 255 accounting for the expansion of these sequences. He also predicted that some, and perhaps most, genes in complex organisms express regulatory RNAs and that the evolution of RNA regulatory networks was the enabler of the appearance and radiation of developmentally complex animals. 217 That is, plant and animal genomes are not full of non-functional remnants of early evolution colonized by parasitic genetic hobos but are largely devoted to the specification of regulatory RNAs required for multicellular development 256–266 (Chapters 12–14 and 16). Later studies showed, inter alia, that ‘enhancers’ with tissue-specific activity (Chapters 14 and 16) are enriched in introns 267 and that many small regulatory ‘microRNAs’ are derived from introns (Chapter 12). 268
Measured in picograms / cell, converted to base pairs on the basis that 1 base pair = 660 daltons, assuming an equimolar amount of the bases, i.e., 1 pg = 10 9 base pairs, or 1,000 Mb (1 Gb).
The definition of biological complexity is controversial and susceptible to pedantry. We define three types of complexity: metabolic complexity, which is collectively high in microorganisms, and lower in plants and animals; developmental complexity, the numbers of positionally and functionally distinct cells and structures (Chapter 15); and cognitive complexity, the ability to process information and learn, which is highest in mammals (Chapter 17).
It should be noted, however, that the smallest amphibian genome is half the size of the smallest mammalian genome. See http://www
Some species and cell types increase chromosomal/chromatid copy number during development. The giant polytene chromosomes of salivary glands in Drosophila is one example.
Upward exceptions do not negate the possibility that large amounts of regulatory DNA are needed to program the ontogeny of complex organisms.
Wheat is a familiar example. 34
Interestingly, based on mutational load arguments at the time, Müller estimated that there would be ~30,000 genes in mammals, repeated by Ohno, which turned out (much later) to be surprisingly accurate for protein-coding genes. 49 King and Jukes used similar calculations to predict an upper limit of 40,000 essential genes. 50 Such considerations do not apply to regulatory sequences if variations within them lead to complex trait variation, shown later by genome-wide association studies (Chapter 11).
The work of Garrod, Cuénot, Beadle and Tatum, Luria and Delbruck, Lederberg, Benzer, Müller and others on ‘biochemical mutations’ that led to the ‘one gene – one enzyme’ hypothesis (Chapter 2).
Which highly influenced early geneticists, including R. A. Fisher (a founder of the field of Population Genetics) and the Modern Synthesis.
These were the first manifestations of the nascent field of bioinformatics, pioneered also by Margaret Dayhoff and Richard Eck, 69 who introduced the concept of molecular phylogeny, reflecting a prescient prediction by Crick a few years earlier, when he stated: “Biologists should realise that before long we shall have a subject which might be called ‘protein taxonomy’—the study of the amino acid sequences of the proteins of an organism and the comparison of them between species. It can be argued that these sequences are the most delicate expression possible of the phenotype of an organism and that vast amounts of evolutionary information may be hidden away within them.” 70
Despite the fact that RNA editing has expanded greatly and the enzymes involved have been subject to strong positive selection in the primate lineage (Chapter 17).
That is not to say, however, that genetic drift is not an important evolutionary process, and there are likely many passenger or hitchhiker sequence variations of subtle effect. 97
These contraforces are hard to disentangle over evolutionary time. By definition, any useful variation is subject to positive selection until it becomes fixed in the population (appearing initially to have evolved rapidly by supplanting the previous sequence). It is then subject to negative selection as its loss is disadvantageous, and thereafter evolves slowly (Chapters 10 and 11).
In general, there are exceptions, such as the ultraconserved elements whose sequences evolved more rapidly than those of proteins during tetrapod evolution but are evolving far more slowly in the amniotes (birds and mammals; Chapter 10).
Recent high-resolution data suggest that transcription factor binding sites occupy just 0.2% of the genome. 153
In 1986, John McCarrey and Art Riggs proposed that pseudogenes might have roles as regulatory switches or ‘determinator-inhibitor pairs’ during development based on antisense relationships, 165 a prediction validated, at least in part 166–169 (Chapters 9 and 13).
Later high-throughput studies showed that a large fraction of protein-coding genes in E. coli and yeast do not result in lethality or in easily discernible phenotypes when deleted, presumably because laboratory conditions do not recapitulate natural selection for more subtle functions or variations in gene expression. 175 , 176 The same problem, of limited phenotypic screens, applies also to animals, especially in relation to inter- and intra-species competition, and behavioral and cognitive characteristics.
Such as the exome sequencing and genome-wide association studies, Chapter 11.
They “acquired the anthropomorphic labels of ‘selfish’ and ‘parasitic’ because of their replicative autonomy and potential for genetic disruption”. 16
It is clear that retroviral and retrotransposon insertions in some instances disrupt protein-coding genes, 185 but it is also clear that some, if not many or most, have far from random genomic- and clade distributions and have been exapted to function, ‘nature’s tools for genetic engineering’ 186–188 (Chapters 10 and 16).
The etymology is exon = EXpressed regiON, coined by Gilbert in 1978: “The notion of the cistron… must be replaced by that of a transcription unit containing regions which will be lost from the mature messenger – which I suggest we call introns (for intragenic regions) – alternating with regions which will be expressed – exons.” 157
Other studies showed that at least 40% of all RNAs in human cells are not polyadenylated (Chapter 13). 221–223
Transcription and translation are tightly coupled in prokaryotes, meaning that introns in protein-coding genes are disruptive, whereas the separation of transcription from translation by the nuclear membrane in eukaryotes allows intron excision before translation. The only introns found in prokaryotes to date are located in rRNA and tRNA genes, which would not be subject to such strong negative selection.
Open Access: This content is Open Access under the Creative Commons license CC-BY-NC-ND.
Your browsing activity is empty.
Activity recording is turned off.
See more...