© 2023 John Mattick and Paulo Amaral.
Open Access: This content is Open Access under the Creative Commons license CC-BY-NC-ND.
Bookshelf ID: NBK595933DOI: 10.1201/9781003109242-14

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mattick J, Amaral P. RNA, the Epicenter of Genetic Information: A new understanding of molecular biology. Abingdon (UK): CRC Press; 2022 Sep 20. doi: 10.1201/9781003109242-14

It had been known for decades that DNA is wound around structural units called nucleosomes in eukaryotic chromatin and that chromatin is variably compacted and remodeled during development. Higher resolution studies from the 1980s showed the existence of chromosome territories, protein-coding gene-rich and -poor regions, and fine-scale ‘topologically associated domains’ with different GC-contents and distributions of sequences derived from transposable elements. Genetic loci termed enhancers, hundreds of thousands of which exist in mammalian genomes, were found to control plant and especially animal development, apparently involving chromatin ‘looping’ to activate protein-coding genes in their vicinity. Nucleosomes were found to contain canonical and specialist histones, some specific to mammalian germ and neuronal cells. Histones were found to be subject to a bewildering variety of post-translational modifications that are imposed, interpreted and erased by protein complexes that have no intrinsic sequence specificity, including many such as Polycomb and Trithorax previously shown to be essential for the developmental regulation of gene expression. Histone modifications vary by gene expression and differentiation state. Exons are preferentially located in nucleosomes, suggesting that epigenetic control of gene expression may be exon specific. Vertebrate DNA is methylated at cytosines in CpG dinucleotides dynamically during development, perturbed in cancer, and associated with heterochromatin formation and gene repression, but little is known of the pathways that determine the locus specificity of DNA and histone modifications during development or in response to environmental parameters.
Early studies in Drosophila and other organisms showed that the patterns of gene expression vary in different cell types, which define their identity and fate, and that these patterns can be maintained following DNA replication and subsequently through mitosis. That is, there is a secondary form of genomically encoded heritable information, termed ‘epigenetic’ information, which is embedded in chromatin modifications and manifested as canalized pathway choices during differentiation and development, first proposed by Conrad Waddington in the 1940s. 1–5
The developmentally regulated packaging of eukaryotic DNA into compacted heterochromatin a and more transcriptionally active euchromatin had been known since the early 20th century, with different regions of the genome thought to be open or closed for business, akin to a library compactus. 8 , 9
In 1974, Ada and Donald Olins 10 and Roger Kornberg 11 , 12 reported that eukaryotic chromatin appears like “linear arrays of spheroid units” or “beads-on-a-string”, respectively, and that the DNA is wound like cotton around a spool into 11 nm diameter ‘nucleosomes’, which contain four pairs of histones. 11 The Olins also credited another investigator, Christopher Woodcock, who had obtained similar images. Woodcock’s paper 13 was, however, rejected by the journal Nature, a reviewer asserting that to accept the article would require “rewriting our textbooks on cytology and genetics” and that “such a naïve paper … should not be published anywhere”. 14
It was known from the 1960s from the work of Vincent Allfrey, Alfred Mirsky and others that histones can be methylated or acetylated, sometimes in response to external stimuli, and that these modifications affect transcription, 15–17 although the extraordinary range of histone modifications was not apparent until much later.
Pluripotent cells have relatively open chromatin, as do cancer cells, whereas the extent of closed chromatin increases as cells differentiate. 8 , 18 Nucleosomes in heterochromatin are compacted into higher-order structures, initially described as 30 nm fibers, but the exact nature of these structures remains controversial. 19–22 Chromatin is further compacted during meiosis and mitosis. 23–26 While the mechanisms controlling chromatin condensation and decondensation are not well understood, it is clear that histone modifications and non-histone proteins play important roles. 27 , 28 Moreover, in all eukaryotes – from yeast to plants and animals – RNAs have been shown to be associated with chromatin, degradation of which by RNase changes the patterns of exposed DNA. 29–31
The fine-scale organization of the eukaryotic nucleus, chromosomes and chromatin becomes more elaborate with increased developmental complexity, documented by Torbjorn Caspersson, Julie Korenberg, Mary Rycowski, Georgio Bernardi, Wendy Bickmore and others, who also showed that cytological ‘banding’ patterns, gene density, intron density, protein density, GC content, CpG island and repeat distributions vary widely across chromosomes. 32–40
The classical banding patterns correlate with the distribution of repeats. In human chromosomes Alu elements are concentrated in the so-called Reverse or R-bands, especially in the T-bands, the most intensely stained and most GC-rich fraction of the R-bands. LINE1 elements are concentrated in the alternating Giemsa or G-bands 33 , 35 , 39–41 and sequester genes with specialized functions in the nucleolus and inactive lamina-associated domains (see below), indicating a global role of transposable elements in orchestrating the function, regulation and expression of their host genes. 42
In situ fluorescent hybridization studies by Thomas and Marion Cremer, Bickmore and others from the 1990s showed that chromosomes occupy defined ‘territories’ in the nuclei of animal and plant cells 43–48 (Figure 14.1), confirming the conclusions drawn by the cytogeneticists Carl Rabi and Theodor Boveri a century before. 49–51 These studies, refined and expanded by new techniques, also revealed radial segregation of chromosomal domains: gene-rich and actively transcribed chromosomal regions are located in the center of the nucleus, whereas gene-poor and genetically quiescent heterochromatic regions are sequestered at the periphery, associated with the nuclear membrane. 47 , 51–56
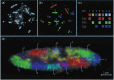
Chromosome territories (CTs) in the chicken fibroblast nucleus. (Reproduced from Cremer and Cremer with permission of Springer Nature.)
Job Dekker and colleagues showed that, in both animals and plants, euchromatic and heterochromatic regions are partitioned into megabase-sized active ‘A’ and inactive ‘B’ compartments, respectively, 57 , 58 which encompass smaller three-dimensional ‘topologically associated domains’, or TADs, with high-frequency intra-chromatin interactions. 22 , 57–71
A striking example is the discovery by Elphège Nora, Dekker, Edith Heard and colleagues that the X-inactivation center (XIC), which they failed for decades to define using cloned transgenes of up to 500 kb, spans bipartite TADs b that occupy ~800 kb of genomic territory: the promoter for the Xist gene, which triggers X inactivation, lies in one TAD of ~500 kb, whereas its antisense regulator Tsix lies in another TAD of ~300 kb. 76 , 77 They also proposed that TADs underlie many properties of the long-range transcriptional regulation that occurs in animals and plants, 61 , 76 a prediction that coalesces with later observations that subnuclear and subcellular compartment organization is at least partly driven by RNA-mediated phase separation 78–82 (Chapter 16). The topological organization of chromatin during development is also reliant on repetitive elements and their interaction with the heterochromatin 1 (HP1) protein family. 41 , 83
TADs have an average size of ~0.5–1 Mb, shown by proximity ligation (cross-linking the DNA in situ to identify sequences physically adjacent in three-dimensional space), 60 with higher resolution analyses revealing finer scale internal TAD organization. 70 , 84 TADs appear to demarcated by boundary regions anchored by the ‘insulator’ protein CTCF c and the ‘cohesin’ complex (Figure 14.2), which interact and control chromatin loop extrusion, 64 , 86–91 involving phase-separation, 92 evident as “architectural stripes”, where loop anchors secure topological domains and link enhancers (see below) to cognate promoters. 90 A cell lineage-specific subset of CTCF binding sites d and TAD boundaries are controlled by DNA methylation, e indicating an interplay between epigenetic modifications, chromatin organization and transcript isoforms during development. 94–100
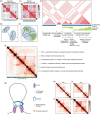
The structural features of topologically associating domains. (a–d) Heat-map representations (top) and schematized globular interactions (bottom) of TADs (a,b) and nested subTADs (c,d). (e) Cartoon representation of different classes of contact (more...)
CTCF is also associated with attachment to the nuclear lamina, a filamentous protein network underlying the nuclear membrane in animal cells, where it demarcates ‘laminar-associated domains’ (LADs). LADs have low transcriptional activity, 101 consistent with the earlier observations that gene-poor and quiescent genomic regions are located at the nuclear periphery. The composition of the nuclear lamina varies in different tissues, and mutations in laminar proteins result in a range of conditions including muscular dystrophies and neurological disorders. 102 Lamin-like proteins also occur in plants and dynamically tether heterochromatin to the nuclear periphery in response to environmental and developmental signals. 55
Vertebrate genomes are also partitioned into ‘isochores’, megabase-sized domains of different G+C content, which are most pronounced in mammals. 103 Isochores may correlate with TADs and LADs, with the G+C distribution apparently playing a role in “moulding” chromatin accessibility, although the relationship is unclear. 104
The number of LADs, TADs and replication domains (~2,000) in the human genome is similar to the number of chromosome bands observed in prometaphase chromosomes. 105 , 106 TADs and TAD boundaries also correspond with the bands and inter-bands seen on Drosophila polytene chromosomes, 107 as well as with ‘chromomeres’ – locally coiled chromatin domains observed in mitotic and meiotic prophase chromosomes, 108 , 109 supported by the observation that TADs are condensed chromatin domains separated by regions of active chromatin. 85
Some reports suggest that TADS are stable across evolution, cell types and independent of gene expression, and may represent DNA replication modules, 8 , 63 , 111–114 whereas others indicate that TADs, and to a lesser extent A and B compartments, vary among cell types and are reorganized during differentiation and development. 48 , 60 , 64 , 71 , 115 TADs may be equivalent to the chromatin domains formed by enhancer action 84 , 116 (see below). TADs in human pluripotent stem cells are demarcated, at least in part, by transcriptionally active HERV-H retrotransposons 117 and regulated by the RNAi pathway via AGO1 association with expressed enhancers. 118 Some evidence suggests that megabase-scale TADs are largely cell-type invariant, whereas ‘subTADs’ reconfigure in a cell type-specific manner. 110 TAD reconfiguration at the HoxD locus appears to regulate limb development, 119 and cell-type specialization is encoded by chromatin topologies. 115
TADs are also reorganized in response to physiological parameters, such as hormone signaling and neuronal activation. 120–122 They are also the functional units of the DNA damage response, required for the one-sided cohesin-mediated loop extrusion of chromatin domains containing the double-strand break-specific histone variant, phosphorylated H2A.X (see below), a process that involves transcription of non-coding RNAs. 123 Mutations affecting TAD boundaries are associated with human developmental disorders and cancers, apparently due to aberrant promoter-enhancer interactions. 124 , 125
‘Enhancers’ are upstream, downstream or intronic non-protein-coding genomic regions in animals and (to a lesser extent 126 ) plants that control developmental cell-type-specific spatiotemporal expression patterns of protein-coding and non-protein-coding genes in their neighborhood, by altering the organization of chromatin. 127–133 Enhancers can be located hundreds of kilobases away from their target genes and are (local) position and orientation-independent. 126 , 134–140
Enhancers were classically recognized and genetically defined by their developmental effects, rather than their biochemical properties or mode of action. Although not described as such, enhancer activity was first observed in the bithorax complex of Drosophila, 141–143 but it was only in the early 1980s that the term was coined to describe the unexpected ability of SV40 viral DNA sequences to increase the expression of a cloned β-globin gene. 144 Many tissue-specific enhancers, often containing repetitive elements similar to those in viral enhancers, f were subsequently identified in mammalian immunoglobulin and globin gene g loci, as well as in the Drosophila bithorax complex and other genes that show restricted expression patterns during development (Figure 14.3), initially using deletion strategies. 134 , 135 , 146–151

Restricted expression patterns of embryonic enhancers at two different developmental stages visualized by lacZ expression in transgenic Drosophila embryos. (Reproduced from Stathopoulos et al. with permission of Elsevier.)
Many enhancers have since been identified by other genetic and bioinformatic approaches, 153–155 the former using insertions of transposons with reporter genes, called ‘enhancer trapping’, in Drosophila, 143 , 152 , 156 , 157 plants 158 and vertebrates. 159 More recently attempts have been made to characterize known enhancers and identify others by genome-wide analysis of the binding positions of presumed signature proteins (the ‘transcriptional co-activators’ P300 and Mediator h ) combined with the presence of correlated histone modifications, 167–171 the presence of nucleosome-depleted regions and/or the expression of ‘enhancer RNAs’ (eRNAs), 172–179 which yield different prediction sets and blur the distinction between enhancer and (protein-coding) gene promoters 127 , 139 , 154 , 180 (see below and Chapter 16).
The appearance of enhancers has been linked to the emergence of animal multicellularity and phenotypic diversity, 181 , 182 neuronal expansion in vertebrates 183 and the recent evolution of primates. 184 Positive selection for nucleotide changes in enhancers has contributed, for example, to the uniquely human aspects of thermoregulation (sweat glands in the skin) 185 and digit and limb patterning, including the increase in size and rotation of the thumb toward the palm for enhanced dexterity. 186 Body plan specification is controlled by multiple enhancers to ensure precise patterns of gene expression. 151 , 187 Clusters of enhancers, such as those at the beta globin locus, but also many others, have been dubbed “super-enhancers”, “stretch enhancers” or “enhancer jungles”. 127 , 188–193 Enhancers also play a role in the etiology of cancer, 127 , 192 , 194 and disruptions of chromatin topological domains cause rewiring of gene-enhancer interactions with pathogenic consequences. 64 , 122 , 131 , 136 , 195 , 196
Enhancers are still incompletely defined, physically and conceptually, 128 , 153 , 154 but have been described as “DNA logic gates”. 197 Mechanistically, enhancers were originally conceived as clusters of transcription factor binding sites that are brought into contact with target protein-coding gene promoters by long-distance DNA looping, a model first proposed by Mark Ptashne to reconcile enhancer function with transcription factor control of gene expression. 198 The persistence and vagaries of the initial interpretation of how enhancers work 124 , 128 , 131 , 136 , 169 , 199 has been referred to by Marc Halfon i as a case of “founder fallacy” and “validation creep”, 154 by no means the first in molecular biology or science generally.
There is good evidence that enhancer action leads to the juxtaposition of distal chromosomal sequences in three-dimensional space, and to consequent transcriptional activation of genes in their orbit. 200 Enhancer-mediated DNA looping may be equivalent to TADs 131 , 201 but enhancers can exert their action across TAD boundaries, which may in turn play a role in mediating formation, reorganization and/or juxtaposition of such domains, 122 , 131 , 132 , 139 , 143 , 157 , 202 , 203 although genome topology and gene expression can be uncoupled. 204 Enhancers also recruit histone-modifying chromatin remodeling proteins, such as the CREB-binding protein (CBP, see below). 166 , 205
However, evidence for the direct interaction of transcription factors bound at enhancers with target protein-coding gene promoters is limited, in some cases contradictory, 206 and intimate contact may be more an enduring presumption than an accurate mechanistic description, 154 especially in view of the fact that enhancers are transcribed in the cells in which they are active. 134 , 135 , 146–150 , 172–179 , 207–209 Indeed, enhancers have many if not all of the characteristics of bona fide genes, including promoters. 210 , 211 Most lncRNAs originate from enhancers 209 , 212 and enhancer RNA production is considered the most reliable indicator of enhancer action. 172–179 How enhancers select their targets is unknown, but likely involves RNA-DNA, RNA-RNA and RNA-protein interactions 213–215 (Chapter 16).
Strikingly, the number of mammalian enhancers, estimated to be in the hundreds of thousands, 130 , 170 , 172 , 180 , 192 , 216–219 far outweighs the number of protein-coding genes, which indicates that distal sequences that regulate developmental expression patterns occupy a much larger fraction of the genome than those constituting the proximal promoters of protein-coding genes.
Partial digestion of exposed DNA in chromatin with micrococcal nuclease yields a ladder of modal DNA lengths in multiples of ~180bp, reflecting 147bp of DNA supercoiled around the outside of the nucleosome core particle and ~35bp of linker DNA between (in mammals), although the average length of the linker sequence varies between species and cell types. 220
There are approximately 30 million nucleosomes in a human cell. 221 Canonical nucleosomes are composed of an octamer of four small, highly basic proteins: histones H2A, H2B, H3 and H4; the central H3-H4 tetramer is sandwiched between two H2A-H2B dimers and the N-terminal tails of the histones, which protrude beyond the DNA shell and are the major sites for post-translational modifications 222–224 (Figure 14.4) (see below). Canonical histones are produced during the replicative S-phase of the cell cycle and are among the most highly conserved proteins in evolution. 225 Interestingly, the genes encoding the canonical (but not the variant) histones are some of the few genes that lack introns, possibly as their constitutive production with chromosomal replication does not require efference signals to be transmitted in parallel.

(a) Nucleosome structure showing histone octamer core, encircling DNA and protruding histone tails. (Reproduced from Luger et al. with permission of Springer Nature.) (b) Some of the many modifications of histone N-terminal tails. (Reproduced from Zhao (more...)
Archaeal histones form a structure similar to the eukaryotic H3-H4 tetramer, but, unlike eukaryotic histones, lack extended N-terminal tails and post-translational modifications. 227 Both possess a copper (Cu2+) binding site at the H3 dimerization interface and have been shown to have copper reductase activity, 228 suggesting that they originated as a mechanism for copper utilization under oxidizing conditions. 229
Another histone, H1, binds to the outside of the nucleosome at the entry and exit sites of the DNA to stabilize the particle and/or play a role in coiling of nucleosomes into higher-order structures. 230 , 231 A homolog of histone H1 exists in bacteria, and also appears to have been acquired by eukaryotes at the time of their origin. 232
Nucleosomes were initially thought to be simply a means of compacting genomes – there is ~2.5 m of DNA in a mammalian cell – and this is likely an important function. Nonetheless, they are not static but dynamic structures, histones being exchanged and differentially modified during differentiation and development. 231 , 233–235
The promoters of protein-coding genes and developmental enhancers initially appeared to be ‘nucleosome-free regions’ based on their sensitivity to DNase digestion and accessibility to transcription factors. 236 However, more sensitive approaches have revealed that nucleosomes do occur in the vicinity of promoters but are “unstable” and subject to higher turnover. 31 , 237–243
There are also variant forms of nucleosomes, mostly involving H2A. H2A can be replaced by H2A.Z, which, unlike other histones, is multi-exonic and produced throughout the cell cycle. H2A.Z is present in most tissues, but most highly expressed in embryos, essential for development in insects, vertebrates and plants 244–248 and associated with memory formation. 249
There exist two H2A.Z genes encoding almost identical proteins (three amino acid differences) in chordates, one of which expresses a primate-specific alternatively spliced isoform in the brain. 250 , 251 The two H2A.Z subtypes display differential occupancy at the promoters of protein-coding genes and enhancers, and regulate genes involved in early embryological, neural crest and craniofacial development development, 246 , 252 as well as the progression of some types of cancers. 253 Just one of the three amino acid differences between the H2A.Z subtypes is sufficient to rescue the developmental abnormalities caused by mutations in an enzyme that catalyzes replacement of the canonical H2A-H2B dimer with the H2A.Z-H2B dimer. 252
The H2A variant H2A.X is recruited to double-stranded DNA breaks and its phosphorylated form is required for their repair, a process that is also involved in programmed genomic rearrangements during immune cell development. 254 , 255
Another H2A variant (‘macroH2A’) contains an additional and highly conserved large C-terminal domain that has homologs in all kingdoms of life and is covalently linked to its N-terminal histone homology domain, which is also highly conserved but quite different from that in the canonical H2A. 256 The macro domain has ADP-ribosylation activity and possibly RNA-binding activity. 257 MacroH2As are encoded by two multi-exonic genes, one of which is alternatively spliced in the macro domain. They associate with the inactive X chromosome of female mammalian cells and inactive genes and appear to have a role in maintaining heterochromatin. 258 , 259
There are also short variants, H2A.B, H2A.L, H2A.P and H2A.Q and splice isoforms thereof, which lack the C-terminal tail of the core H2A. These variants appeared in mammals and are tissue-specific, being expressed in the testis and, in the case of H2A.B, also in the brain. 260 The H2A.B variant binds RNA, replaces H2A.Z in nucleosomes at transcription start sites and intron-exon boundaries in the testis and the brain, and interacts with RNA polymerase II to promote the activation of transcription. 261–263 It is also involved in biparental inheritance controlling embryonic development in mice. 264 H2A.B has a propensity for chromatin decompaction 261 , 265 and co-localizes with the RNAi proteins Miwi and Dicer in spermatids, 260 indicating a relationship between regulatory RNAs, chromatin organization and splicing pathways.
The H2A.L.2 variant also has an RNA-binding domain and appears to be guided to its sites of incorporation by RNA. 266 In sperm development it dimerizes with the H2B testis-specific variant TH2B as a prelude to nucleosome displacement by other highly basic proteins called protamines, 260 originally discovered by Miescher, 267 which mediate the extreme compaction of the chromosomes.
Histone H3 is replaced in nucleosomes by H3.3 (which differs from H3 by only four amino acids) in telomeres and pericentromeric regions and when chromatin assembly occurs at times other than replication, 268–272 including in meiotic sex chromosome inactivation. 273 Histone H2A-H2B is bound to an essential telomerase RNA domain, which suggests a role for histones in the folding and function of the telomerase RNA component. 274
H3.3/H2A.Z double variant-containing nucleosomes are enriched in active promoters and enhancers. 237 , 238 Loss of H3.3 results in fertility and/or defects in gastrulation or neural crest development in flies, fish and frogs. 237 , 275–277 In mammals, H3.3 also accumulates in neurons, reaching near saturation by adolescence, where it controls neuronal- and glial-specific gene expression patterns, with an essential role in plasticity and cognition. 278 Rare missense mutations in H3.3 have been shown to cause neurologic dysfunction and congenital anomalies. 279 Mutations of lysines in the tail of H3.3 are commonly observed in glioblastomas. 280 , 281
In flies and vertebrates, there are two seemingly redundant genes encoding H3.3, H3A j and H3B, which vary in their 3′UTRs, 272 whose individual loss in mammals causes infertility and reduced viability. 283 , 284 Loss of both causes embryonic lethality, due to heterochromatic dysfunction at telomeres and centromeres, 285 the latter of which can be rescued by injecting dsRNA derived from pericentromeric transcripts, indicating a functional link with the silencing of such regions by an RNAi pathway. 284
In centromeres, H3 is replaced by another variant, CENP-A, which is essential for kinetochore formation required for chromosome segregation during mitosis and meiosis. 286 , 287 In plants, epigenetic memory is reset by replacing H3 with the variant H3.10 (which is refractory to lysine 27 methylation) during sperm maturation to globally reprogram paternal gametes. 288
We could go on. The bottom line is that there is a high degree of complexity in the composition of nucleosomes, dynamic histone exchange and remodeling of chromatin during development. 289 However, little is known about the decisional processes and mechanisms that determine when and where different histones are incorporated into particular nucleosomes, other than that RNA and ‘pioneer transcription factors’ are involved (Chapter 16).
Pioneer or ‘architectural’ transcription factors, such as Sox2 and Sox11, k which are ‘high mobility group’ (HMG) proteins, bend DNA structure and initiate the opening of chromatin by eviction of the linker histone H1. 290–293 Other pioneer factors, such as the winged helix/forkhead box (Fox) proteins, bind to DNA within nucleosomes in promoters and enhancers leading to their destabilization, also by histone H1 displacement, and recruit Mediator and cohesin to permit chromatin access for tissue-specific remodeling factors such as FoxA, with different targets in different cells at different stages of development. 294–298
Histones are escorted to nucleosomes by companion or ‘chaperone’ proteins, 268 , 286 , 299 , 300 and histone exchange requires a conserved family of ATP-dependent ‘chromatin remodeling enzymes’ variously known as SWI/SNF, ISWI, NuRD, CHD and INO80, 301 many now known to be regulated by cis- and trans-acting non-coding RNAs (Chapter 16).
Histone modification by methylation and acetylation was first observed and proposed to have a regulatory function in the 1960s, and nucleosomes were known to affect transcription, 302–304 but it was not until 1991 that Michael Grunstein and colleagues provided definitive evidence of gene regulation by histone acetylation. 305 In 1996, David Allis and colleagues isolated a histone acetyl transferase, making use of the fact that histones in the macronucleus of Tetrahymena cells are highly acetylated whereas those in the micronuclei are not, which for the first time directly linked a transcriptional regulator to a histone-modifying enzyme. 306 , 307 A reciprocal histone deacetylase (HDAC) activity was reported a month later by Stuart Schreiber and colleagues. 308
Shortly thereafter it was shown that the mammalian ‘transcriptional co-activators’ CBP, P300 and the yeast ‘transcriptional adaptor protein’ Gcn5 function in multi-subunit complexes to acetylate histones in nucleosomes. 160 , 309 , 310 linking what had been vaguely referred to as ‘transcription factors’ to chromatin modification. These findings changed the perception of the nucleosome from being simply a mechanism for genome compaction to a major player in regulating its expression.
The 1990s and 2000s saw the identification of a bewildering array of histone modifications, mainly by mass spectrometry – many of which still remain to be characterized 311 , 312 – at last count in over 60 different positions, mainly in the N-terminal tails of the histones, which are intrinsically disordered 313 , 314 (Chapter 16) and exposed beyond the periphery of the nucleosome. l These modifications span mono-, di- and tri-methylation, acetylation, ADP ribosylation, ubiquitylation and/or sumoylation of various lysines in histones H2A, H2A.X, H2B, H3 and H4, mono- and di-methylation, acetylation and deimination of arginines (to citrulline m ) in H2A, H3 and H4, phosphorylation of serines, threonines, tyrosines and one lysine in H2A, H2A.X, H2B, H3 and H4, isomerization of prolines in H3, and O-palmitoylation of a serine in H4. 27 , 320 , 321
Histone modifications also include propionylation, butyrylation, malonylation, formylation, glutathionylation, tyrosine hydroxylation and lysine crotonylation, 311 , 312 , 322 , 323 the latter at 28 different lysines in H1, H2A, H2B, H3 and H4. 324 Many of these modifications are in low abundance, suggesting particular contextual functions. An example of their impact, however, is that citrullination of histone H1 leads to its displacement from the nucleosome and the decondensation of chromatin in pluripotent cells and during developmental reprogramming. 316 , 322
The shorthand nomenclature for modifications is histone > amino acid (single letter code) > position > modification – for example, the methylation of arginine 11 on histone H4 is written as H4R11me, and the acetylation of lysine 5 on histone H2B is written as H2BK5ac, etc.
Other more exotic modifications have been discovered, such as histone ufmylation (the conjugation of UFM1 ubiquitin-like protein to H4 to promote DNA repair), 325 , 326 the covalent conjugation of the metabolite lactate at 28 sites on core histones (histone “lactylation”) 327 and the conjugation of the neurotransmitters serotonin and dopamine to H3 glutamine 5 (H3Q5ser) and trimethylated lysine 4 (H3K4me3Q5ser) in specific regions of the brain 328 , 329 Cocaine administration, which causes dopamine release from the ventral tegmental area (VTA), induces hyperacetylation n of H3 and H4 at genes associated with cocaine addiction in the nucleus accumbens, a brain ‘reward’ region. 331 Moreover, rats undergoing withdrawal from cocaine dopaminylate histone H3 glutamine 5 (H3Q5dop) in the VTA, inhibition of which reverses cocaine-mediated gene expression changes, attenuates dopamine release in the nucleus accumbens, and reduces cocaine-seeking behavior. 328 These are potentially profound observations for understanding brain function – neurotransmitters have lasting epigenetic effects.
There are over 100 enzymes known to catalyze histone modifications at particular amino acid positions in mammals (called code ‘writers’), and dozens more that remove them (‘erasers’), o mostly acting on histone H3, 332 with a similar albeit less extensive repertoire in other animals, plants and fungi. Many of these proteins are encoded by homologs of genes first identified as critical for Drosophila development, notably Polycomb, Trithorax and Zeste (Chapter 5). The two multi-subunit Polycomb complexes in mammals, PRC1 and PRC2, act non-redundantly at target genes to maintain transcriptional programs and cellular identity. PRC2 methylates lysine 27 on histone H3 (H3K27me), while PRC1 ubiquitinates histone H2A at lysine 119 (H2AK119ub), 333 both preferentially at unmethylated CpG islands, 334 with a complex interplay between them, including, for example, with the core PRC component EED, which recruits histone deacetylases. 335 , 336 Trithorax proteins, which activate gene expression, contain the SET domain, which methylates H3K4 and is found in all eukaryotes. 337
Substantial innovations in the subunit composition of chromatin-modifying complexes have accompanied increased developmental complexity. Histone modification writer, reader and eraser complexes are more elaborate and diverse in mammals than invertebrates. The Drosophila PRC1 complex, for example, has just one version of its constituent subunits, whereas mammalian PRC1 can incorporate any one of two RING subunits, three PHC subunits, six PCGF subunits and five CBX subunits, 338–340 the latter of which interact with the neural gene repression factor REST 341 and appear to be involved in the formation of local phase-separated domains 342 (Chapter 16).
Similar increases in subunit complexity and/or the numbers of orthologs or genomic binding sites also occurred in the Mediator complex in metazoans, 162 , 343 CTCF in bilaterians, 344 the HUSH complex for heterochromatin regulation in vertebrates, 345 and the major expansions of the fast evolving zinc-finger transcription factors (one of the largest gene families in humans), many of which have associated metazoan-specific BTB, tetrapod-specific KRAB or mammal-specific SCAN domains. 346 , 347
Different types of histone modifications are recognized by over 70 known ‘reader’ proteins, many of which contain Tudor, PHD finger, MBT, bromo or chromo domains that occur in a range of chromatin remodeling and histone-modifying factors. 332 , 348–352 PHD fingers read the tail of histone H3, primarily the methylation state of H3K4 (K4me3/2), and to a lesser extent the methylation state of H3R2 (R2me2) p and the acetylation state of H3K14. 352 Bromo domains q primarily recognize acetylated lysine residues, 356 and occur along with acetyltransferase domains in the pioneer factors CBP and P300. 357 Chromo, Tudor and MBT domains are part of an extended family that evolved from a common ancestor and recognize methylated lysines. 358 , 359
Underscoring their importance, mutations in histone modification writers, readers and erasers cause developmental abnormalities, intellectual disabilities and cancers. 360–364 For example, 10% of leukemias are caused by translocations and ectopic fusions of the Trithorax homolog KMT2A (lysine-specific methyltransferase 2A), previously called MLL1 – for ‘mixed lineage leukemia’ 1. 365 Dysregulation of the chromatin-binding PHD finger protein JARID1, which binds H3K4me2/3, also causes leukemias. 366 Haploinsufficiency of histone deacetylase 4 (HDAC4) results in brachydactyly mental retardation syndrome. 367 A number of drugs that inhibit histone deacetylases have been licensed for use against hematopoietic cancers, particularly lymphomas and myelomas. 368
In 2000, David Allis and Brian Strahl proposed the ‘histone code hypothesis’:
First, the establishment of … a combinatorial pattern of histone modification, i.e., the histone code, in a given cellular or developmental context … Second, the specific interpretation or the ‘reading’ of the histone code … (which) function broadly to set up an epigenetic landscape that determines cell fate decision-making during embryogenesis and development. 370
The last sentence is the key and far-reaching conclusion, which takes gene regulation in eukaryotes well beyond conventional transcription factors and suggests that epigenetic processes comprise the senior level of control of developmental trajectories, notwithstanding the fact that the differentiation state of cells can be changed by ectopic expression of transcription factors (Chapter 15).
It has taken a long time for this view of the regulation of cell fate during development to overcome the hegemony of transcription factors, and there has been staunch opposition to it. As Allis later recalled:
Chromatin studies in this era paled in comparison with the more exciting studies on transacting transcription factors that were all the rage … Moreover, well defined paradigms of gene regulation had been elegantly worked out in prokaryotic models … Histone proteins were viewed as only being in the way of where all of this exciting action took place. My career choice to study histone biology was a steep uphill climb, especially given the popular notion that histones did not really matter in gene regulation. 371
Even after histone modifications were shown to have a role in the regulation of the expression of iconic genes involved in development, their action was widely interpreted in terms of nucleosome control of transcription factor accessibility, rather than considering what might regulate nucleosome position and histone modification state in the first place.
The emphasis on transcription initiation as the main focus of ‘gene regulation’ and the resistance to the suggestion that epigenetic regulation may determine which genes are available to be transcribed are perhaps best illustrated by a 2013 article by Mark Ptashne, who pioneered the characterization of transcription factor binding to DNA in bacteria and yeast. 198 , 372–374 Ptashne’s article, entitled ‘Epigenetics: Core Misconcept’, 375 stated:
Development of an organism from a fertilized egg is driven primarily by the actions of regulatory proteins called transcription factors … Rather, it is said, chemical modifications to DNA … and to histones … drive gene regulation. This obviously cannot be true because the enzymes that impose such modifications lack the essential specificity … and so these enzymes would have no way, on their own, of specifying which genes to regulate under any given set of conditions. 375
The latter point is correct, but Ptashne and many others overlooked the possibility that the specificity might be supplied by trans-acting RNAs, despite the fact that he had elsewhere recognized that RNA molecules can act as a transcriptional co-activators. 376 Of course, regulation of chromatin organization and transcription initiation is not mutually exclusive nor separable; the factors involved act in concert to govern the complex patterns of gene expression during development (Chapter 15).
Deciphering the histone code is a huge challenge, not the least because of the difficulty of analyzing the modifications and their effects on gene expression at the nucleosome level, the dependency of the context of the large combination possibilities of chromatin marks, and the heterogeneity of the samples. Nonetheless, the growing popularity of the field not only led to the rapid discovery of the many enzymes and complexes involved 377 , 378 but also the roles of modifications by a number of pioneering labs, r using in vitro approaches (e.g., with reconstituted nucleosomes) and modification-specific antibodies for global analysis of the in vivo distribution of nucleosomes containing the modification. 27 , 320 , 379–381
The latter revealed non-random patterns of modifications in different tissues and developmental stages, such as in the Neurod2 gene in the brain (Figure 14.5), hypoacetylation of the inactive X chromosome in female mammals and silent mating type genes in yeast, and hyperacetylation of the upregulated X chromosome in Drosophila males or transcribed globin genes in erythrocytes. 381
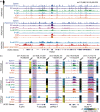
Dynamic landscape of histone modifications at the mouse Neuro2d (Neuronal Differentiation 2) locus in different tissues and during development. (Reproduced from ENCODE Project Consortium under Creative Commons CC BY license.)
High-resolution mapping by sequencing of immunoprecipitated chromatin (‘ChIP-seq’) has shown that histone modifications are differently imposed in complex patterns at millions of different genomic positions in different tissues or cell types at different stages of differentiation and development. 382–386 There is clearly also ‘crosstalk’ between histone modifications, 387–389 which may occur in modules, but little is yet understood of the lexicon or syntax. 384
Active genes are characterized by acetylation of various lysines or arginines, which neutralizes their charge interactions and makes chromatin more accessible. 368 , 390 Different acetylations are found in different regions of genes and regulatory regions: H2AK9ac, H2BK5ac, H3K9ac, H3K18ac, H3K27ac, H3K36ac and H4K91ac are mainly located in the region surrounding the transcription start site, whereas H2BK12ac, H2BK20ac, H2BK120ac, H3K4ac, H4K5ac, H4K8ac, H4K12ac and H4K16ac are elevated in the promoter and transcribed regions of active genes. 384
Nonetheless, even the roles of well-studied modifications, including acetylation, of different histones and residues by distinct complexes in different cell types and species are far from fully characterized. For example, in human cells the histone acetyltransferase KAT8 s modifies different H4 residues (H4K5 and H4K8 vs H4K16) depending on its associated proteins, with different regulatory and pleiotropic effects. 394 H3K27ac marks are generally thought to distinguish active enhancers from inactive/poised enhancers that contain H3K4me1 alone, 167 , 395 although H3K27ac alone is insufficient to permit enhancer activity 396 (see below).
Conversely trimethylation of the same lysine (H3K27me3) marks facultative heterochromatin (regions that are differentially expressed in development and/or differentiation), such as the inactive X chromosome. 397 This modification is imposed by PRC2 through one of two alternative catalytic subunits, EZH1 or EZH2, t which are expressed at different stages of development. 398 Mutations in EZH2 cause Weaver Syndrome, which is characterized by skeletal and cognitive abnormities. 399 Mutually exclusive acetylation and methylation also occur at other lysines including H2BK5, H3K4, H3K9 and H3K36, all of which are acetylated at active promoters. 384
H3K27me3 has been widely implicated in restraining the expression of lineage-specifying and cell-state defining loci from plants to animals, 288 , 400–403 and mutation of this residue recapitulates PRC2 transformations. 404 Its role in regulating the timing of the differentiation of progenitor cells has also been linked with epigenetic switches controlled by opposing PRC2 and Kdm6a/b demethylase activities, for example in regulating T cell commitment timing in mammals. 405 H3K27me3 repression of gene expression also appears to be confined within TADs. 402 , 406
There have been various attempts to use signature histone marks to identify enhancers, with initial correlations with the binding of the transcriptional co-activator P300 suggesting that enhancers are characterized by the presence of monomethylated histone H3 lysine 4 (H3K4me1) and the absence of trimethylated H3K4 (H3K4me3). 170 However, subsequent studies showed that H3K4me3 is enriched, whereas H3K4me1 is reduced, in highly active enhancers, 169 , 407 and that characterized enhancer regions contain a variety of histone modifications in different combinations, not necessarily the presumed canonical H3K4me1 or H3K27ac marks. 171 , 384 , 408 , 409 Bioinformatic predictions of enhancers based on histone modification patterns alone have low validation rates. 155 , 407 , 410
The H3K4me3 modification is not only associated with active enhancers but also with actively transcribed protein-coding genes, 411 or genes ‘poised’ u for transcriptional activation. 415–417 H3K4me3-modified histones exhibit a peak around transcriptional start sites 390 and interact with RNA polymerase subunit TFIID. 349 , 418–420 Transcription start sites also exhibit a typical flanking bimodal pattern of H3K4me2- and H3K4me3-marked nucleosomes. 420 , 421
So-called ‘bivalent domains’ containing both H3K4 and H3K27 methylation occur around conserved non-coding sequences associated with developmentally important transcription factors, suggesting that chromatin state is important for maintaining embryonic pluripotency. 422 , 423 Recent data shows that pluripotent states are determined by interactions between chromatin modifications and enhancer expression to reconfigure the target specificity of the pioneer transcription factors Oct4, Sox2 and Nanog. 424 Other modifications, such as H4K16ac occur in active enhancers and protein-coding genes, 171 further obscuring the distinction between them.
H3K14pr and H3K14bu are (also) preferentially enriched at promoters of active genes 323 and H2AK119ub1 guides maternal inheritance and zygotic deposition of H3K27me3 in mouse embryos. 425 , 426 Histone H4 lysine 16 acetylation (H4K16ac), a hallmark of decondensed, transcriptionally permissive chromatin, directly stimulates the Dot1 histone H3K79 methyltransferase. 389 H3.3 variants are phosphorylated at S31 in gene bodies for high-level activation of rapidly induced genes, shown in macrophages to be coordinated with SETD2 methylation of H3K36 to effect recruitment and ejection of chromatin regulators. 427
Constitutive heterochromatin in genomic regions such as the centromeres and telomeres contain high levels of H3K20me3 and H3K9me3, 398 , 417 the latter of which binds the repressive HP1 protein via its chromodomain. 348 , 428 H3K4me3 and H3K9me3 mark imprinting control regions. 417 Histone sumoylation appears to act as a repressive mark by recruiting HDACs to gene promoters, 429 and H3K36me is present in nucleosomes along the body of transcribed genes, and is necessary for efficient constitutive pre-mRNA splicing by recruiting the chromo domain protein Eaf3 to mediate interaction with the splicing machinery. 430 , 431
These are just some examples. The patterns are complex and studies are becoming more sophisticated. 369 Targeted deposition or removal of histone modifications using CRISPR/Cas9-fusions and related approaches, such as single-cell CRISPR screens and chromatin modification profiling by mass spectrometry, are starting to allow the dissection of causal roles for individual modifications. 432–439
An important discovery was that nucleosomes are preferentially positioned over exons, 440–443 suggesting that histone modifications convey exon-specific information, and that epigenetic control of gene expression extends to the level of individual exons. This offers a mechanistic explanation for the observed coupling of chromatin structure, transcription and splicing, 444 including the physical co-location of alternatively spliced exons with promoters, 445 and a basis for exon selection by histone modifications at different stages of development in different cell types and different conditions, 443 , 446–453 which appears to be controlled in part by small RNAs. 454–456
Chromatin-modifying proteins have a profound impact on developmental processes because they lie at the functional center of epigenetic regulatory networks. They do not make (although they do convey) locus-specific regulatory decisions but rather are directed by other information that does. How histone modification writers and erasers select particular nucleosomes at particular genomic positions for particular modifications in different cell types is unknown, but is likely RNA guided (Chapter 16). The histone modifications and nucleosome positioning must be tightly controlled during development, as developmental trajectories are precise (Chapter 15), although histone modifications are also influenced by metabolic and physiological factors. 457–460 Moreover, histone-modifying proteins are themselves subject to post-translational modifications. 461 , 462 which suggests yet more layers of developmental control and environmental tuning.
Histone modifications are often inherited through meiosis and mitosis, to transmit information between generations 425 , 426 , 463 , 464 (Chapter 17) and to ‘bookmark’ loci for reactivation or maintenance of heterochromatin after cell division. 465–467 The available evidence is that the parental core H3-H4 tetramer is split and segregated strand-specifically at the replication fork, that parental histones are recycled to sister chromatids and re-incorporated near their original positions, maintaining their acetylation and methylation marks, possibly asymmetrically to alter cell fate in daughter cells. 468 , 469
Replication timing maintains the global epigenetic state in human cells. 470 It is still unclear, however, how the histone modifications are inherited, particularly in view of the report that epigenetic memory is independent of symmetric histone inheritance replication, 471 although histone-modifying enzymes remain associated with DNA during replication. 472 , 473 Epigenetic marks are also erased and reset with every round of transcription (which involves similar disassembly of or navigation through nucleosomes), 469 , 473–476 again possibly involving RNA direction. 234 , 353 , 477
The imposition and maintenance of this information clearly involves histone modification writers but this does not explain their locus specificity, which probably operates to exon level. Whatever the mechanism(s), the amount of information involved, and stored in the genome, must be enormous.
All four bases of DNA are subject to modifications, more than 20 of which have been identified, 478 , 479 the most common being methylation of cytosines and adenosines. In bacteria, v DNA methylation (mainly m6A) is used to protect endogenous sequences against restriction endonuclease cleavage (Chapter 6), but also has roles in DNA replication and gene expression. 481–484 Adenosine methylation has been reported in protists, plants and animals, 485–492 although emerging evidence suggests that the source may be microbial or RNA contamination. 493 , 494
5-Methylcytosine (m5C) occurs widely in eukaryotic genomes and is the best studied. In fungi and plants, cytosine methylation is used to silence viruses and transposons, as well as to regulate development 495–497 and environmental responses, 498 , 499 likely related processes. In maize, the cycling of transposable elements between active and inactive states to regulate local gene expression is determined by the methylation state of the element, 495 , 500 , 501 which may also be in part the role of TEs in animals.
Most invertebrate genomes are not heavily methylated and some species such as Drosophila and C. elegans appear to lack DNA methylation, indicating that it does not play a role in their development, although it is used for genome defense and gene regulation in other invertebrates. 502 , 503 For as yet unknown reasons, a major evolutionary transition from fractional to global methylation occurred at the origin of the vertebrates, 504 as did the appearance of regional variation in GC content. 103
DNA methylation as a major player in gene regulation in mammals first came to light in the 1970s with the observation that there is differential methylation of the mammalian X chromosomes. 505 , 506 Later studies showed that there is widespread erasure of methylation in the mammalian germ line w and in early development, 513–516 selective reimposition of methylation at different loci including enhancers in different cell lineages, 497 , 516–518 and reactivation of genes (and induction of tumors) by a cytosine analog (5-aza-cytidine) that cannot be methylated. 519–521 Embryonic stem cells maintain their pluripotent state in the absence of DNA methylation, but cannot differentiate. 496
In mammals, methylation is primarily, but not solely, associated with repression of gene expression, notably in inactivated X chromosomes, x pericentromeric heterochromatin, imprinted loci and the regulation of transposons (which are related, as most of the targets of methylation are TE-derived) and occurs mostly and symmetrically in cytosines in CpG dinucleotides, except those clustered in so-called ‘CpG islands’. 523–528 Deamination of methylcytosine yields thymidine, which is thought to account for the underrepresentation of CpG dinucleotides in mammalian genomes. 523 , 529
The sequence symmetry of CpG enables propagation of the methylation mark through cell division, which combined with its complex interplay with Polycomb repressive and other histone-modifying complexes 530 , 531 and its differential patterns during development, led to the proposal that CpG methylation comprises a pathway for cellular memory of transcriptional states. 505 , 506
CpG islands occur mainly in promoters (including those of enhancers 219 ), especially those of broadly expressed housekeeping genes, 529 , 532 , 533 methylation of which correlates negatively with gene expression, although repression of these promoters appears to occur primarily by H3K27me3 histone modifications. 496 , 531 , 534 Genes with CpG island promoters also have other characteristic epigenetic signatures, including high levels of H4K20me1, H2BK5me1 and H3K79me1/2/3 at their 5′ end. 535
In contrast, tissue-specific protein-coding genes usually, but not always, lack islands. 529 , 532 , 533 In transcriptionally active genes, CpG islands are devoid of methylation and enriched for permissive nucleosome modifications such as H3K4 methylation. On the other hand, DNA methylation is enriched in the body of highly transcribed genes, often associated with H3K36 methylation, 430 , 535–538 where it influences nucleosome positioning 539 and alternative splicing, 540 phenomena that may be linked. It has been recently shown that hypomethylated CpG dinucleotides preserve an archive of tissue-specific developmental enhancers in adult mouse cells, marking decommissioned sites and enabling recovery of epigenetic memory, 541 a process involving the pioneering factor FoxA and TET2/3 methylcytosine dioxygenases 542 (see below).
Cytosine methylation is carried out by DNA methyltransferases, of which vertebrates have three: two ‘establishment’ DNA methylases (Dnmt3a and 3b), and one ‘maintenance’ methylase (Dnmt1) that recognizes hemi-methylated CpG sites following DNA replication. All three are required for embryonic development, with mutations causing syndromic developmental neurological, sensory and immunological defects, and loss of Dnmt1 in neurons at later stages resulting in cognitive defects. 361 , 543–545 The histone mark H3K36me2 also recruits Dnmt3a to regulate intergenic DNA methylation 537 and H3K23 ubiquitylation couples maintenance DNA methylation with replication. 546 While DNA methylation is thought to be stable, it is cycled at promoters at high frequency, suggesting an updating mechanism. 547 , 548
The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation, 530 and is essential for brain development and function. 549 , 550 Loss of MeCP2, which is encoded on the X chromosome, causes a neurological disorder called Rett Syndrome with variable penetrance in females (due to variable patterns of X inactivation) whereas its loss in males usually leads to severe congenital encephalopathies and early death. 550 Dnmt3a and MeCP2 originated at the onset of vertebrates, with methylation of non-CpG sites being exceptionally high in the mammalian brain and regulating highly conserved developmental genes, with a likely role in the evolution of cognition. 551
Dnmt2 was originally thought to be a DNA methyltransferase but is, in fact, a tRNA methyltransferase, and it seems likely that the modern DNA methyltransferases evolved from an ancient RNA methyltransferase. 552–554
Methylcytosine is converted to hydroxymethylcytosine (hmC) by TET proteins, 555 which can also further oxidize hmC to generate 5-formylcytosine and 5-carboxylcytosine. 556 There are three TET proteins in mammals with different expression patterns and different targets during development. 557 , 558 TET proteins hydroxymethylate DNA at enhancers and telomeres, 559 and TET1 and TET2 associate with Nanog to facilitate reprogramming of somatic cells to pluripotency. 560–562 Formation of 5-hMC is required in embryonal stem cells for the maintenance of pluripotency and inner cell mass specification. 557 It is also required in the brain, especially in Purkinje cells, where it is almost 40% as abundant as meC. 563 TET3 is present in neurons and oligodendrocytes but absent in astrocytes. 564 TET3 regulates behavioral adaptation in the neocortex, 565 as well as synaptic transmission and plasticity in the hippocampus, 566 and its loss results in increased anxiety-like behavior and impaired spatial orientation. 564 Fear extinction, an important form of reversal learning, leads to a dramatic genome-wide redistribution of 5-hmC within the infralimbic prefrontal cortex, and learning-induced accumulation of 5-hmC is associated with the establishment of epigenetic states that promote gene expression and rapid behavioral adaptation. 565
DNA methylation patterns have been extensively analyzed following the discovery by Marianne Frommer and colleagues that bisulfite treatment of DNA converts cytosine, but not meC, to uracil, which sequences as T, 567 , 568 and more recently by direct DNA sequencing using nanopore technology, which can distinguish modified from unmodified bases. 569 , 570 For this reason (technical ease of analysis) and its earlier discovery, DNA methylation has been more widely studied than histone modifications, notably in the ‘Human Epigenome Project’, which revealed differences in methylation patterns in different cell types and interplay between genetic variations and epigenetic state during development and aging, y in the brain, in cancer and other diseases such as arthritis 226 , 545 , 572–575 (Figure 14.6). Akin to the insights now routinely offered by RNA-seq, new techniques will increasingly reveal the variety and dynamics of epigenetic states and transcription factor occupancy at single-cell resolution during development and in diseases such as cancer. 576 , 577
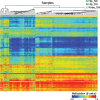
Aberrant methylation patterns in enhancer loci in cartilage chondrocytes from patients with hip osteoarthritis (OA) and knee OA, compared to healthy controls (NC). (Reproduced with permission from Lin et al. under Creative Commons CC BY license.)
However, as with histone modifications, there is little known about the signaling pathways that direct the locus-specific imposition or removal of cytosine methylation by generic enzymes during development, learning and disease, except that the RNAi pathway is involved (Chapter 16).
The roles of the (admittedly at the time vague) organization of chromatin in the regulation of gene expression, and the mechanisms that might be involved, were rarely considered when the bacterial model was extrapolated to developmentally complex eukaryotes. Accordingly, since then, the regulation of gene activity by chromatin architecture has been viewed predominantly through the lens of DNA-binding transcription factors.
This interpretative lens led to the loose and confusing description of many proteins that are required to mediate the patterns of gene expression during development, such as those that organize chromosomal domains or modify chromatin, as ‘pioneer transcription factors’ or ‘transcriptional co-activators’, 304 , 578–581 despite the fact that they have no intrinsic or only vague DNA-binding specificity (Chapter 16). The implied assumption biased the interpretations of experimental observations and retarded the understanding of the control of gene expression during development by placing proteins that are required to instruct genome architecture into the same conceptual and mechanistic basket as those that bind to specific sequences to activate or inhibit transcriptional initiation.
Appreciating that chromatin modifications play a central role in the regulation of gene expression during development has also been confused by the term ‘epigenetic inheritance’, implying that it is separate from ‘genetic’ (DNA-based) inheritance, obscuring the fact that the unfolding cascade of epigenetic modifications must be instructed by information that is encoded in the genome.
The key challenges are to consider how much information is required to orchestrate organismal ontogeny (Chapter 15) and to identify the pathways that connect chromatin modifications, enhancers, effector proteins and other layers of genome regulation during ontogeny (Chapter 16). How this information is modulated by the environment and during learning is addressed in Chapter 17.
There are two types of heterochromatin: facultative heterochromatin, which is developmentally regulated (such as occurs in X-chromosome inactivation and at many other discrete loci during differentiation and development), and constitutive heterochromatin (such as occurs in centromeric and telomeric regions of chromosomes). 6 , 7
The structure of these domains on the X chromosome and others on autosomes is regulated by the lncRNAs Dxz4 and Firre. 72–75
There is also conflicting data, with other studies showing a poor correlation between CTCF binding sites and TAD boundaries. 85
Many CTCF binding sites are derived from transposable elements. 93
DNA methylation also regulates alternative polyadenylation via CTCF and the cohesin complex. 94
Endogenous retroviruses have been shown to be a source of enhancers. 145
Globin enhancers were originally and are still often referred to as ‘locus control regions’. 146
P300 is a histone modifying enzyme. 160 Mediator is a highly modular multi-subunit complex that appears to connect distal transcription factors with the transcription initiation machinery. 161–163 Both P300 and Mediator bind RNA, which is required for their chromosomal localization, TAD juxtaposition and local chromatin modification 164–166 (Chapter 16).
Halfon notes, for example, that “a recent paper erroneously states that enhancers ‘were first described as nucleosome-depleted regions with a high density of sequence motifs recognized by DNA-binding transcription factors’.” 154
The coding sequence of H3A appears to have evolved under strong purifying selection in the lobe finned fish and tetrapods, without any change to the amino acid sequence. 282
Sox2 and Sox11 are involved in the maintenance of pluripotency and neuronal differentiation, respectively.
Some modifications also occur in the internal globular domains of the histones. 311
Citrulline is also an intermediate in the urea cycle. Citrullination catalyzed by peptidylarginine deiminases (PADs) neutralizes arginine’s positive charge, can antagonize arginine methylation for local gene regulation and global chromatin decompaction, with implications for cell pluripotency and differentiation. 315 , 316 The peptidylarginine deiminase PAD4 is essential for the remarkable formation of neutrophil extracellular (chromatin) traps (NETs) that are phagocytosed by macrophages to stimulate innate immune responses during infection. 317–319
Alcohol consumption also increases histone acetylation in fetal and adult mouse brain. 330
The discovery of histone modification erasers was unexpected by many, as were the discoveries of DNA demethylases (see below) and RNA modification erasers (Chapter 17). Indeed, most levels of regulation beyond transcription factors were initially met with skepticism, and then largely shoehorned into the transcription factor paradigm.
Bromodomain proteins have been explored as a target for anticancer drugs, with mixed results. 355
Including those of Allis, Shelley Berger, Rudi Jaenisch, Thomas Jenuwein, Manolis Kellis, Tony Kouzarides, Bob Kingston, Danny Reinberg, Bing Ren, Bryan Turner, Rick Young, Jerry Workman, Shi Yang and many others.
KAT8, also known as MOF or MYST1, is classically associated with H4K16 acetylation in transcriptional activation, notably in the MSL complex that executes the roX RNA-directed global upregulation of the expression of the X-chromosome in Drosophila males for dosage compensation. Disruption of the orthologous human MSL complex also impairs H4K16 acetylation and results in an X-linked syndrome marked by developmental delay, gait disturbance and facial dysmorphism, 391 as well as tumor maintenance by exacerbating chromosomal instability, 392 , 393 the latter exemplifying that histone modifications have other roles in chromosome biology beyond the regulation of gene expression.
EZH1 and EZH2 contain the lysine-specific SET (Su(var)3-9, Enhancer of Zeste, Trithorax) domain that uses the cofactor S-adenosyl-L-methionine (SAM) as the methyl donor.
Bacterial DNA can also be modified by phosphorothioation, as part of an alternative restriction-modification defense system. 480
The spreading of X-inactivation from the ‘X-inactivation center’ on the X chromosome in females appears to be mediated by methylation of LINE elements distributed along the chromosome. 522
The link between genetic variations and epigenetic state of regulatory elements affecting gene and trait expression is illustrated by the classic example of lactase non-persistence in mammals and the selection for lactase persistence during aging observed in many Europeans and other pastoral cultures, which involves non-coding variations, a specific lncRNA (LOC100507600 or Lactase antisense RNA 1), RNA interference and DNA methylation in intronic enhancers. 435 , 571
Open Access: This content is Open Access under the Creative Commons license CC-BY-NC-ND.
Your browsing activity is empty.
Activity recording is turned off.
See more...