© 2023 John Mattick and Paulo Amaral.
Open Access: This content is Open Access under the Creative Commons license CC-BY-NC-ND.
Bookshelf ID: NBK595931DOI: 10.1201/9781003109242-10

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mattick J, Amaral P. RNA, the Epicenter of Genetic Information: A new understanding of molecular biology. Abingdon (UK): CRC Press; 2022 Sep 20. doi: 10.1201/9781003109242-10

By the mid-1990s, the extraordinary advances in DNA cloning, amplification and sequencing had made feasible the mapping and sequencing of whole genomes. The exponential growth of data led to extensive and progressively well-annotated genome databases, and suites of computational tools for gene prediction, homolog identification and gene structural and expression analyses. For the first time the full complement of DNA sequence information in bacteria, simple eukaryotes, fungi, plants and animals began to be revealed, enabling comparative genomics to interrogate evolutionary relationships and functional indices at increasingly high resolution. Prokaryote genomes were confirmed to be dominated by protein-coding genes, with phenotypic diversity achieved primarily by proteomic variation. On the other hand, animals differing by orders of magnitude in developmental complexity were found to have a similar number and repertoire of protein-coding genes - only about 20,000 in both nematodes and mammals - the ‘G-value enigma. By contrast, increased developmental complexity correlated with the extent of intronic and intergenic non-protein-coding DNA, indicating that phenotypic radiation and developmental sophistication in multicellular organisms is achieved mainly by regulatory expansion. Mysterious non-protein-coding ‘ultraconserved’ sequences and large numbers of ‘pseudogenes’ were found in mammalian genomes. Different classes of transposable element and retroviral-derived sequences were characterized in plant and animal genomes and shown to be major drivers of phenotypic innovation.
The prelude to genome sequencing was genome mapping, physically for microbial genomes, which generally range from 400 kb to ~10 Mb, and, initially, genetically for animal and plant genomes, which are orders of magnitude larger (Chapter 7).
Physical mapping was performed by cleavage of genomic DNA with restriction endonucleases with rare recognition sites and electrophoretic size separation of the resulting fragments, which were then ordered into linear or circular maps by partial or sequential digestion and DNA cross-hybridization. Genetic markers were integrated into these maps in well-studied bacterial species, such as E. coli (4.7 Mb) 1 and Pseudomonas aeruginosa (5.9 Mb), 2 as well as in the brewing and baking yeast Saccharomyces cerevisiae, which also has a relatively small genome enabling relatively accurate estimation of its size. 3 Maps were also developed for other well-studied fungi, notably the ‘fission’ yeast Schizosaccharomyces pombe (which is also used in brewing a ) and Neurospora crassa.
Genetic mapping of large genomes based on the frequency of co-inheritance of linked markers was pioneered in Drosophila in the early part of the 20th century (Chapter 2) and well advanced by its end. Genome maps based on linkage analysis were also constructed for other widely studied species, such as maize, rodents, cattle and the model flowering plant Arabidopsis thaliana, in which mutants were first described in 1873 (see 4 ) and which gained wide currency from the 1950s and 1960s, especially when it became obvious that, by plant standards, it has an unusually compact genome.
However, physical mapping of genomes of complex organisms at high resolution was a monumental task beyond the capability of any laboratory or consortium before the 1980s. The restriction fragment pattern was far too complex for any individual segment to be resolved and identified – a blur on electrophoretic display – until these genomes could be partitioned by cloning.
In the 1970s, Hogness, Welcome Bender and colleagues constructed libraries of randomly cloned large inserts that encompassed the entire Drosophila genome (Chapter 6). This allowed physical restriction enzyme maps to be developed for individual segments and genomes to be virtually assembled by ‘chromosomal walking’ across overlapping segments. It also allowed the screening of these libraries for specific sequences by ‘colony hybridization’ and the first ‘positional cloning’ of a gene, Ultrabithorax, by mapping a disruptive inversion, an approach that was then extended to many other alleles and genes in which mutants had arisen by chromosomal breakages or transposon insertions. 5–9
In 1980, Christiane Nüsslein-Volhard and Eric Wieschaus undertook systematic genome-wide screens for genes involved in Drosophila development, 10 which led to the discovery of the components of the major signaling pathways, many of which then turned out, like Ultrabithorax, Polycomb and Trithorax, to have homologs in other animals, including mammals. These new genes were then isolated and characterized by positional cloning and transposon tagging, 11 , 12 an approach extended to other species including Arabidopsis and mice.
Similar large insert libraries and physical maps of chromosomes were developed for many other organisms and used extensively for the mapping of mutations, especially those causing human genetic disorders, using restriction site polymorphisms as guideposts to track alleles in affected families (Chapter 11). They were also used as the platforms for whole genome sequencing projects prior to the introduction of highly parallel random sequencing and assembly approaches.
In the 1980s, Martin Evans, Oliver Smithies and Mario Capecchi developed methods for constructing transgenic mice using retroviral vectors and homologous recombination in embryonal stem cells, 13–15 which permitted the introduction of specific mutations to examine their consequences and the rescue of mutant phenotypes by gene transfer. 16–21 These approaches were further extended by ‘enhancer traps’ to screen for genes based on their pattern of expression 22 and systems for ectopic expression. 23
While some viral and organelle genomes had been sequenced, b the sequencing of organismal genomes was made feasible by the development in the mid-1980s of fluorescently labeled oligonucleotide sequencing primers by Leroy Hood and colleagues, which enabled optical (laser) reading of electrophoretic displays of fragments generated by the Sanger chain termination method and the consequent development of highly parallel automated DNA sequencers 26 (Chapter 6).
Using this technology, the first whole genome from an organism to be sequenced was that of the bacterium Haemophilus influenza by Craig Venter, Hamilton Smith and colleagues in 1995, 27 who devised a strategy of ‘shotgun’ cloning and sequencing to avoid the tedious work of mapping a genome and the difficulty of coordinating laboratories working on different parts of it. Venter and Smith rationalized that it was easier, at least for small genomes, to sequence random fragments en masse, and then assemble a continuous sequence in silico by matching overlaps, called ‘contigs’, c a logical extension of their shotgun sequencing of human cDNAs. 28
And so it proved, and the sequence of 1.83 Mb H. influenzae genome was completed 2 years before that of E. coli (albeit having a larger genome, 4.6 Mb 29 ), whose sequencing was begun earlier. This was the first time that molecular biologists were able read the entire genome sequence of a living cell, identify all of the protein-coding genes (Figure 10.1) and start to understand its genetic programming and evolutionary history holistically, exemplified by the insights gained from sequencing the intracellular parasite Rickettsia prowazekii, the causative agent of epidemic typhus. 30
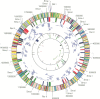
Circular map of the H. influenzae genome illustrating the location of key restriction sites (outer perimeter), color-coded predicted coding regions (outer concentric circle), regions of high and low G+C content (inner concentric circle), coverage of clones (more...)
These studies revealed many previously unknown features of bacterial genomes, including remnants of bacteriophages – which proved the signpost to a spectacular new technology for genetic engineering (Chapter 12) – and other sequences that suggested genome evolution and plasticity through transposition and horizontal DNA exchange, often using bacteriophages as the vehicle.
Thousands of bacterial and archaeal genomes have since been sequenced and deposited in the public databases. The data reveal that prokaryotic genomes, while encoding some short regulatory RNAs (Chapter 9), are comprised, in the main, of protein-coding genes, separated by short regions that contain cis-acting transcriptional and translational control sequences. d Nonetheless, their genomes collectively encode extraordinary proteomic diversity and fluidity of gene content, reflecting their range of ecologies from commensal pathogens to deep ocean volcanic vents and industrial waste. 35 , 36
For example, most E. coli strains contain between 4,000 and 5,000 genes, e but only 20% of the genes in a typical E. coli genome are shared among all strains, 37 which is the core proteome that defines the species, whereas the total number of different protein-coding genes observed in different strains exceeds 16,000. 34 , 38 A recent analysis of 303 million bacterial genes from 13,174 publicly available metagenomes showed that most genes are specific to a single habitat and that the majority of species-level genes and protein families are rare. 39 That is, phenotypic diversity in prokaryotes, primarily metabolic and ecological versatility, is achieved by varying the proteome.
The first eukaryotic genome to be sequenced was the 12.4 Mb genome f of S. cerevisiae, achieved chromosome-by-chromosome by an international consortium led by André Goffeau in 1996, which identified almost 6,000 protein-coding genes and 140 rRNAs, 40 snRNAs and 275 tRNAs. 41 , 42
The S. cerevisiae genome contains only a few (~270) short introns (average 247bp), located in just 4.5% of protein-coding genes, 43 deletion of which was later shown to have physiological and growth effects, 44–47 i.e., even these small introns contain information. Interestingly, the number of introns in S. cerevisiae is substantially lower than in the superficially similar S. pombe, whose 13.8 Mb genome has fewer protein-coding genes (~4,900) but many more introns (~4,700, in 40% of protein-coding genes), albeit still mainly small (30–800bp, modal length 48bp). 48
Both S. cerevisiae and S. pombe have proven invaluable in the genetic dissection of the control and mechanics of cell division and other basic processes such as protein trafficking, and the identification of homologous genes in plants and animals, including human. 49 , 50 Indeed, genome analyses showed that the protein components of core cellular processes have been conserved throughout eukaryotic evolution.
Large numbers of yeasts (including such pathogens as Candida albicans, which causes thrush) have now been sequenced, showing, for example, that many brewing yeasts are hybrids of S. cerevisiae and S. eubayanus, and that both originated in East Asia, with different strains being domesticated in different places. 51 , 52
The genome sequence of N. crassa was published in 2003 and found to contain ~10,000 protein-coding genes, with introns and intergenic regions occupying ~56%. About 10% of the genome is comprised of repetitive sequences. 53
And, of course, the genomes of important parasites, such as Plasmodium falciparum, which causes malaria, were also soon sequenced, 54 along with that of its mosquito host in 2002. 55 The richness of the information in these genome sequences is extraordinary, and still being investigated. To do so has required the construction and maintenance of databases and the acquisition of bioinformatic tools and skills, a major change to the investigative landscape of all biological domains, from evolution to neurobiology.
The first (nearly) complete sequence of the genome of any multicellular organism was that of C. elegans, accomplished in 1998 by a consortium led by John Sulston, 56 who (with Sydney Brenner and H. Robert Horvitz) pioneered it as an experimental organism. 57 , 58 C. elegans has only ~ 1,000 somatic cells, whose ontogeny had been determined, and has been a useful model for many processes including cell differentiation (Chapter 15), RNA interference (Chapter 12), transgenerational inheritance (Chapter 17), drug responses such as nicotine withdrawal 59 , 60 and aging, 61 among others. The C. elegans genome was found to be 97 Mb in size and to contain just over 20,000 protein-coding genes, “one-fifth to one-third the number predicted for humans”. 56 Seventy-three percent of the C. elegans genome is comprised of introns (26%) and ‘intergenic’ (47%) sequences (Figure 10.2).

C. elegans and map of its genome: Distributions of predicted genes (pale blue); EST matches (green); yeast protein similarities (dark blue); and inverted (purple), tandem (red), and TTAGGC repeats (black) along each chromosome. Numbers are Mb. (Reproduced (more...)
Two years later, the sequence of the Drosophila melanogaster genome was completed by a consortium led by Venter and Jerry Rubin. 62 This time the approach was different, not sequencing of previously mapped cloned segments, as was the case with C. elegans, but mainly sequencing of random fragments, as had been done with H. influenzae. This achievement put paid to the skepticism that many had expressed of this approach because of the problem of repetitive sequences in genome assembly; shotgun sequencing is now the standard method.
The Drosophila genome is ~120 Mb in length and encodes only ~13,600 protein-coding genes, many of which have equivalents in humans, 63 only twice the number of protein-coding genes in yeast and fewer than in C. elegans, which is developmentally far simpler than an insect. 62 This anomaly was noted in a commentary at the time: “… there is little relationship between total gene number, neuron number, morphology and behavioral capacities of diverse organisms in different phyla … (which) merely highlight our ignorance of biological complexity and how it is instantiated.” 64
By contrast, the Drosophila genome contains a greater proportion than C. elegans of introns (>41,000 ranging up to 70 kb in length, for example, in the bithorax and DMD g genes) and ‘intergenic’ sequences, which collectively comprise ~80% of the genome, one of the first hints from genome sequencing that increased developmental complexity is not a function of the number of protein-coding genes, but rather of information in non-coding regions.
In the same year (2000), the first plant genome sequence was also published, that of Arabidopsis thaliana, which has one of the most compact plant genomes known (125 Mb), similar in size to that of Drosophila, but contains almost twice as many protein-coding genes, ~25,500. 68 The genome sequences of two rice cultivars (~450 Mb; 30–50,000 protein-coding genes) were published in 2002. 69 , 70
A ‘first draft’ of human genome sequence was published in 2001, 71 , 72 and a more complete compendium in 2004 73 (Chapter 11), revealing the full extent of the complement of sequences derived from transposable elements, other repeats, introns and ‘intergenic’ regions. The mouse genome was published in 2002, 74 followed soon by the sequences of rat, 75 dog, 76 cow, 77 chimpanzee, 78 chicken 79 pufferfish, 80 the ascidian Ciona (a primitive chordate) 81 and many others.
These studies revealed high conservation of the protein-coding gene complement among vertebrates (~20,000 protein-coding genes, 75% orthologous between fish and human), and especially mammals (~90% orthologous), 82 with lineage-specific expansion or contraction of some gene families such as those encoding cytokines and olfactory receptors. 83–85
The genome of the pufferfish (Takifugu ruprides, often referred to simply as Fugu) was sequenced because it is unusually compact (just 365 Mb, an order of magnitude smaller than in human, but three times bigger than in Arabidopsis), and held up as a model of a streamlined vertebrate genome with minimal ‘junk’. The Fugu genome contains 11% protein-coding, 22% intronic and 67% intergenic non-coding DNA, 17% comprised of repetitive sequences. 80
The unexpected finding from the genome projects was the lack of correlation between the number of protein-coding genes and developmental complexity. 86 Up until this point, gene number had been widely proffered to be a valid measure of biological complexity 87 – and may still be, if the definition of a ‘gene’ is extended to those encoding regulatory RNAs (Chapters 12, 13 and 16).
To recap, C. elegans, a simple nematode with only ~1000 somatic cells has ~20,000 protein-coding genes, as has its sister species C. briggsiae. 56 , 88–90 Sponges, the most basal metazoans, have ~30,000 protein-coding genes. 91 The far more complex insect Drosophila has ~13,600 protein-coding genes, mosquitos have ~16,000, 55 , 92 whereas the water flea Daphnia has ~30,000, the increase in the latter apparently related to ecological flexibility rather than developmental complexity. 93
Humans have ~40 trillion cells sculpted into a myriad of different muscles, bones and organs with complex architectures, 94 as well as a brain with approximately 85 billion neurons (Chapter 15), 94 , 95 but just ~20,000 protein-coding genes, similar to C. elegans and other mammals. 96–102 Indeed, despite fluctuations, the number of protein-coding genes remains remarkably static across the animal kingdom, despite enormous differences in developmental complexity and cognitive capacity. 103 , 104 Moreover, the majority of protein-coding genes in animals are orthologous, including most of those involved in multicellular development and brain function. 91 , 105 That is, all animals have a similar protein toolkit.
On the other hand, in contrast to lack of scaling of protein-coding genes, the fraction of the genome that is intronic and ‘intergenic’ increases with developmental complexity, crudely defined as the number of different ‘cell types’ (Chapter 7), 106–108 although this definition underestimates the different spatial identities, architectures and ontogenies of functionally similar (e.g., muscle or bone) cells (Chapter 15). Prokaryotes have ~10%–15% non-protein-coding sequences, mainly specifying cis-regulatory elements controlling transcription and translation. The non-coding fraction of the genomes of unicellular eukaryotes (protists) generally lies in the range of 40%–50%, fungi 50%–60%, plants 70%–90%, and animals mostly in excess of 90%, with the human genome having 98.8% non-coding DNA (Figure 10.3). 103 , 104
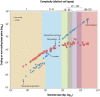
The relationship between biological complexity and genome composition. The y-axis shows the amount of protein-coding sequence (red) and non-protein-coding sequence (blue), which together comprise the total genome size (x-axis) in 76 organisms across the (more...)
Clearly the majority of the information that orchestrates developmental programs and phenotypic diversity lies in the non-protein-coding regions of the genome, which raises the questions of what form the information takes and how is it transduced? The conventional view has been that it involves the combinatorics of cis-regulatory protein-binding sites, more complex post-translational modifications, and expansion of the range of protein isoforms by alternate splicing, h all of which requires additional regulatory information 86 , 113 , 114 (Chapter 15). However, cis-regulatory elements cannot conceivably occupy more than a small fraction of gigabase-sized vertebrate genomes (recently estimated to be ~7% 115 ). On the other hand, the high-throughput RNA sequencing that followed on the heels of genome sequencing revealed that the non-coding regions of animal and plant genomes express thousands of regulatory RNAs in different cells and tissues at different developmental stages (Chapters 12 and 13).
Since that time, there has been a myriad of comparative analyses of genomes. An early example was the comparison of 12 genomes in Drosophila phylogeny, which identified, among other things, many putatively non-neutral changes in protein-coding genes, non-coding RNA genes and cis-regulatory regions, high conservation of shared microRNA sequences, including target mismatches, and adaptive evolution of lineage-restricted microRNAs 116 (Chapter 12). i
Another unexpected finding of the comparison of vertebrate genomes was the discovery of several hundred “ultraconserved” elements >200bp (UCEs) that are identical between the human, mouse and rat genomes, none of which are protein-coding. 118 A subsequent analysis requiring identity of sequences >100bp between any three of five mammalian genomes (human, rat, mouse, dog and cattle) identified almost 14,000 such UCEs, 119 the vast majority of which are not protein-coding, and showed that they evolved rapidly, presumably under positive selection, between fish and amniotes, but then became essentially frozen, subject to fierce negative selection in birds and mammals. 119 , 120
Each UCE is different but has followed the same evolutionary trajectory and so presumably there is some commonality of function. At least some are derived from retrotransposons. 121 They are far more conserved than those specifying protein-coding sequences and rRNAs, which are highly constrained by structure and multilateral RNA-RNA and RNA-protein interactions. Many are enriched in the vicinity of developmental genes and appear to overlap developmental ‘enhancers’ (Chapters 14 and 16) especially in the brain, and many are transcribed into non-protein-coding RNAs, with highly specific expression patterns that are perturbed in cancers and other diseases. 121–131 UCEs are also dosage-sensitive. 132 , 133
However, in contrast to their extraordinary pan-amniote conservation, deletion of four UCEs that function as enhancers in transgenic assays showed no overt developmental perturbation 134 and insertion of sequences into UCEs made no change to enhancer activity, 123 although cognitive phenotypes were not examined. Subsequent deletion of UCEs in the vicinity of the neuronal transcription factor Arx also resulted in viable and fertile mice, but showed subtle neurological or growth abnormalities. 135 Recent results show that the ultraconservation of enhancers is not necessary for their function, 136 and the reason for the fierce conservation of UCEs in birds and mammals remains a mystery. 137
Reciprocally, comparative analyses also identified many RNA genes that have been subject to positive selection in hominid evolution. 138 , 139 One of the most rapidly evolving sequences in the human genome lies within a gene (HAR1F) specifying a highly structured non-protein-coding RNA expressed in Cajal–Retzius neurons during embryonic development of the neocortex, 138 a six-layered structure that is far larger and more complex in humans than in other mammals, including Old World monkeys. 140 , 141 Only two nucleotide changes have occurred in the 118bp HAR1 sequence between chickens and chimpanzees, but there have been 18 changes in the human sequence since our split from the latter. 138 A number of such “human accelerated regions” regulate dosage-sensitive neural genes, acting as enhancers and/or expressing regulatory RNAs, mutations in which disrupt cognition and social behavior. 142–145 Moreover, many primate-specific RNAs, including ‘repeat-derived’ long non-coding RNAs, are involved in a variety of developmental, physiological and cognitive processes (see below and Chapter 13). 146–156
Large numbers of ‘pseudogenes’, rivaling the number of protein-coding genes, were also identified in genomic data – almost 20,000 in the human genome. 157 Pseudogenes are fragments of duplicated protein-coding genes and ‘processed’ intronless copies of mRNAs that (presumably) have been reverse transcribed and retroposed into the genome (‘retrogenes’), which have been interpreted as non-functional ‘molecular fossils’, because they contain incomplete open reading frames or disabling mutations 158–160 (Chapter 7). Curiously, retrogenes are found mainly in mammals. 158 , 161 At least some have been subject to evolutionary selection. 162–164 Many are transcribed in specific cells, and several have been shown to regulate the expression of their protein-coding counterparts, with medical implications (Chapter 13). 165–175
The genome sequencing projects also revealed the repertoire, distribution, age, activity and features of sequences derived from transposons and retroviruses, collectively referred to as TEs (transposable elements), the dominant components of most plant and animal genomes. TEs comprise only a small fraction of yeast, slime mold and Drosophila genomes, can be almost absent or occupy a large fraction of protozoan parasite genomes, and are highly variable both in extent and type in vertebrate and plant genomes (Figure 10.4). 176 , 177
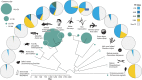
Distribution of TEs across eukaryote phylogeny. Reference genome size (sea green circles) varies dramatically across eukaryotes and is loosely correlated with TE content. Abbreviations: LINE, long interspersed nuclear element; LTR, long terminal repeat; (more...)
In agreement with Britten’s early estimates, nearly half, and perhaps as much as two-thirds, of the human genome is derived from DNA transposons and from short and long interspersed retrotransposable elements (‘SINEs’ and ‘LINEs’) and endogenous retroviruses (‘ERVs’) that replicate and invade genomic sites via RNA intermediates, j although most are now quiescent. 71 , 74 , 179–184 A quarter of these TEs correspond to ~ 1.2 million highly similar, but not identical, copies of Alu SINE elements 67 (derived from 7SL RNA, Chapter 8) that entered the human lineage in three waves during primate evolution, 179 , 180 and were the substrate for a massive expansion of RNA editing, especially in the brain, 185–190 (Chapter 17), with evidence that at least some have been exapted as cell type specific enhancers 191 (Chapters 14 and 15).
Similar numbers and distributions of SINEs (some also descended from 7SL RNA) occur in the mouse genome, although they are distinct from Alu elements and entered the rodent lineage independently. 74 In both species SINEs are clustered in gene-rich regions, especially near promoters, while LINEs (17% of the genome 183 ) are concentrated in “gene-poor” regions and depleted from promoters, 192 indicating different roles (examples in Chapter 16). There are hundreds of thousands of LINE elements in mammalian genomes, but much lower numbers in most non-mammalian vertebrates, 193 although there are exceptions (see below).
The Consortium human genome paper concluded “the organization of Alu elements … suggests that there may be strong selection in favour of preferential retention of Alu elements in GC-rich regions and that these ‘selfish’ elements may benefit their human hosts”, 71 a conclusion confirmed by a later study that showed “that Alu and B1 elements have been selectively retained in the upstream and intronic regions of genes belonging to specific functional classes … (with) no evidence for selective loss of these elements in any functional class”. 194
Indeed, while sequences derived from and distributed by transposable elements have been thought to be largely non-functional (Chapter 7), there is a wealth of evidence, dating back to McClintock’s studies showing transposition altering phenotype in maize and the regulated expression of TEs in development observed by Britten, Davidson and others (Chapter 5), as well as logic, 195 , 196 that TEs are major sources of genetic innovation. 177 , 197–201 They contain and mobilize modular cassettes of (mainly) regulatory information to influence phenotype in evolutionary historical 202–216 and real time. 206 , 217–221 They are perhaps the most important mediators of genetic fluidity, often called ‘jumping genes’ although most do not fit the traditional concept of a ‘gene’, as McClintock intuited by referring to them as ‘controlling elements’ (Chapters 2 and 5). The electronic analogy is control packets.
Thousands of human TEs appear to have undergone positive selection in the vicinity of developmental genes. 222 Other genomic regions, mainly non-coding but also associated with developmental regulation, have been refractory to transposon insertions. 223 A substantial fraction of regulatory sequences in humans, including 25% of promoters and many developmental enhancers, contain sequences derived from TEs. 224 30%–40% of mouse and human RNA transcripts initiate within repetitive elements, 225 , 226 and analysis of approximately 250,000 retrotransposon-derived transcription start sites showed that the derived transcripts are generally tissue-specific, coincide with gene-dense regions and often function as alternative promoters and/or express non-coding RNAs (Chapter 13). 225 Some ancient TEs in the vertebrate lineage contain subsequences that have been retained over huge evolutionary distances. 121 , 227–230
TEs have been shown to be the source of protein-coding and non-coding genes or exons, 121 , 214 , 231–237 centromeres, 238 , 239 transcription factors, their binding sites and networks, 201 , 240–242 lineage-specific regulatory RNAs and tissue-specific developmental enhancers (Chapters 14 and 16), 121 , 153 , 155 , 200 , 224 , 237 , 243–246 promoters and transcription start sites, 200 , 208 , 218 , 225 , 237 , 246–251 epigenetic control modules, 252–259 neocentromeres, 258 targets for parental imprinting, 260 splice sites, 204 , 261 translational controls, 262 microRNAs and microRNA targets, RNA nuclear localization signals 263 , 264 and behavioral modifiers. 265
TEs are the building blocks for epigenetic regulation and chromatin organization, 192 , 255 , 266–269 the senior level of the control of gene expression and cell fate decisions during development in complex organisms (Chapter 14). Many ‘repeats’ are involved in the formation of heterochromatin, the importance of which was historically downplayed, albeit with exceptions 270 (Chapter 7) but now known to be regulated, inter alia, by KRAB zinc finger proteins that bind to TEs 271 , 272 and other transcription factors 273 that have evolved to regulate TE-derived regulatory sequences during embryogenesis and neuronal differentiation 271 , 274 (Chapters 14 and 17).
TEs are also a common source of functional domains in regulatory RNAs, 214 for example, as modules for protein-binding and interaction partners for enhancer action (Chapter 16). They are also prevalent in mRNAs of rapidly evolving mammalian-specific genes. 240 Retrotransposon-derived sequences are widely incorporated into coding and non-coding transcripts in human pluripotent stem cells. 275 Primate-specific retroviral ‘enhancers’ (Chapter 14) and associated TE-containing non-coding RNAs are required for maintenance of stem cell identity and the pluripotency network in humans, 153 , 245 , 276 , 277 and the majority of primate-specific regulatory sequences are derived from transposable elements. 278 They also occur in the most abundant transcripts in the mouse oocyte and regulate gene expression during early embryogenesis. 217 , 279 Numerous retrotransposons act as preimplantation-specific gene regulatory elements and a mouse-specific retrotransposon is essential for mouse preimplantation development. 280 Developmental transitions and cellular stresses increase the expression of both human and mouse SINE transcripts, suggesting a role in both development and physiology. 281–291 LINE1 elements are spliced into non-canonical transcript variants to regulate T cell quiescence and exhaustion. 292 A retrotransposon is also required for small-RNA-induced pathogen avoidance memory in C. elegans and horizontal transfer of that memory to naïve animals. 293
TEs underpin many aspects of quantitative trait variation, due to their capacity to alter gene expression patterns in differentiation and development, and thereby to act as drivers of adaptive/regulatory evolution. 211 Bursts of retrotransposition have been linked with major diversification and speciation events. 213 , 294 Transposon insertions have been associated with developmental innovations and transitions in vertebrates, 200 , 295 including tetrapod evolution, 296 tail loss in the apes, 297 human-specific hippocampal development, 298 the derivation of small breeds of dogs from gray wolves 299 and the differences between Poodles, Boxers and Great Danes. 300 , 301 The ‘calico’ white coat color with spotting in cats arose through a retroviral insertion in an intron that regulates the spatial expression of the c-kit gene, which in turn controls melanocyte differentiation. 302 Similarly, a transposon-derived inverted repeat in an intron of a gene, ‘goldentouch’, is commonly associated with color polymorphism in Midas cichlid fishes. 303
The Rag recombinase proteins involved in V(D)J recombination and the signal sequences therein in the adaptive immune system of vertebrates are also derived from transposons, 304 , 305 as are the regulatory networks underlying MHC (major histocompatibility complex) expression. 306 The regulation of innate immunity has also occurred through the co-option of endogenous retroviruses. 307
The classic textbook example of adaptive microevolution of the British peppered moth into a black form during the industrial evolution, k which was widely cited during the development of mathematical evolutionary theory and the Modern Synthesis (Chapter 2), 308 , 309 proved to be due to an intronic TE insertion that increases the expression of the gene cortex, 310 a member of a conserved family of cell cycle regulators that controls pigmentation pattern, 311 estimated to have occurred in or around 1819, when Charles Darwin was 10 years old (Figure 10.5). 312

Adaptive evolution by transposon insertion into the first intron of the cortex gene of the British Peppered Moth (Biston betularia; panel A) in the early Industrial Revolution, which increases the expression the gene to create the sooty black form (Biston (more...)
Transposon insertions also underlie morphological variations of tomatoes 313 and the changes in the branching structure l that marked the domestication of maize from its wild teosinte ancestor, 314 as well as subsequent flowering time adaptations that allowed cultivars to be grown at higher latitudes. 315 Transposable elements change the color of grapes 316 and apples 317 by insertions in the promoters of genes encoding transcriptional activators of pigment production. Analogous insertions occurred independently in Sicilian and Chinese strains of ‘blood’ oranges, where the cold dependency of the pigmentation reflects the induction of the retroelement by stress. 318 TE mobilization also appears to be a major generator of genetic variation in Arabidopsis. 319
The huge genome sizes of many native and cultivated plants, including wheat, maize, apples and onions that have been selected in recent history, may reflect their greater flexibility to use TE insertions and polyploidy to generate phenotypic plasticity, 215 , 317 , 320–323 in the face of being rooted to the spot, unlike animals, which can move and (consequently) have more precise developmental requirements and limited re-wiring options. This may also explain the extraordinary diversity of phenotypes among closely related plants, such as the varieties produced by artificial selection of the wild mustard plant Brassica oleracea, including broccoli, broccolini, Brussels sprouts, white and red cabbage, cauliflower, kale, and kohlrabi, all of which are the same species. Some animal lineages too – including salamanders and other chordates – may have life history (and specific niche/environmental factors) affecting and being affected by changes in TE content and genome size, with potentially significant impact in adaptations and diversity within the lineage. 324–326
TE insertions are linked intimately to ‘epigenetic’ control of gene activity, notably by methylation 259 , 327 (Chapter 14). The cycling of transposable elements between active and inactive states in maize is determined by the methylation state of the element 328–330 and genome sequencing has revealed the spontaneous insertion of a methylation-insensitive TE-derived ‘epiallele’ in an inbred strain of mouse. 331 Indeed, careful analysis of the features of TEs, which comprise greater than 85% of the maize genome, their insertion sites, expression and methylation profiles, etc., “reveal a diversity of survival strategies … with each TE family representing the evolution of a distinct ecological niche … (and whose impact) is highly family- and context-dependent”. 323
As Nina Fedoroff said in her 2012 Presidential Address to the American Association for the Advancement of Science:
I contend that it is precisely the elaboration of epigenetic mechanisms from their prokaryotic origins as suppressors of genetic exchanges that underlies both the genome expansion and the proliferation of TEs characteristic of higher eukaryotes. This is the inverse of the prevailing view that epigenetic mechanisms evolved to control the disruptive potential of TEs. The evidence that TEs shape eukaryotic genomes is by now incontrovertible. My thesis, then, is that TEs and the transposases they encode underlie the evolvability of higher eukaryotes’ massive, messy genomes… 181
As the genomes of more and more species, including those representing key phylogenetic transitions, are sequenced, and awareness grows, the focus has changed from analyzing the repertoire of protein-coding genes to the nature and distribution of TEs. As a prime example, the publication reporting the marsupial opossum (Monodelphis domestica) genome sequence emphasized innovations in TEs and other non-coding sequences in the mammalian lineage, in sharp contrast with the stasis in protein-coding sequences, as “an important creative force in mammalian evolution”. 332
The 5 Gb genome of Tuatara, the only remaining member of an archaic order that last shared a common ancestor with other reptiles about 250 million years ago and is a link to the now-extinct stem reptiles from which dinosaurs, modern reptiles, birds and mammals evolved, is 64% composed of an amalgam of TEs with both reptilian and mammalian features. 333 In 2021, the complete sequence of the 43 Gb genome of lungfish, the closest living relative of the tetrapods, which is 14 times larger than the human genome, showed that it is 90% composed of intergenic and intronic TEs, mainly LINE elements, that resemble those of tetrapods more than those of ray-finned fish. 296 , 334
Even ‘simple’ repeats (dinucleotide and trinucleotide ‘microsatellites’) or ‘short tandem repeats’, used as markers in gene mapping and DNA fingerprinting, 335 , 336 have been shown to play a role in adaptive radiation, 337 be flexible 338 and function in modulating gene expression. 339 , 340 Simple repeats are also associated with quantitative trait variation, 341 environmental adaptation 342 and human neurodegenerative and neuropsychiatric conditions, 339 , 343 , 344 likely with intergenerational consequences (Chapter 17). The naïve idea that ‘repetitive’ sequences can be a priori and collectively dismissed as junk (with a few ‘exceptions’) is unsustainable in the face of these observations.
A blind spot in genome analysis and comparative genomics, particularly with short-read sequencing, is the difficulty of mapping repetitive sequences and segmental duplications. A related issue has been the widespread use of the ‘RepeatMasker’ program, which masks repeats and low complexity DNA sequences, hiding over 50% of the human genomic sequence. 345 , 346 These problems are being relieved by advent of long-read technologies (see below and Chapter 11), such as nanopore sequencing, which drags single molecules of DNA (or RNA) through engineered protein pores embedded in membranes and measures the disturbance in the electrical current as nucleotides pass through, enabling sequencing of much longer fragments than SBS (over 1 Mb) and direct sequencing of RNA. 322 , 347–355
The pace of genomic exploration was given a huge boost with new technologies allowing massive parallelization of the sequencing process. The most successful to date has been the ‘sequencing by synthesis’ (SBS) method, invented by Shankar Subramanian and David Klenerman, 356 and later commercialized. SBS uses fluorescently labeled nucleotides containing reversible terminators to optically sequence high density clusters of PCR-amplified fragments on solid surfaces. SBS, along with other technologies, permitted a hyper-exponential increase in the volume of DNA sequence data produced and reciprocal reduction in cost – at a much faster rate than the so-called Moore’s Law of computing (at one point a ~2-fold increase in capacity / processing speed and reciprocal halving of cost every 18 months) – the fastest technology revolution in human history. 357
Over the past decade, there has been an explosion of genome sequencing across the entire phylogenetic spectrum (Figure 10.6). Not only have the genomes of tens of thousands of bacterial and archaeal species been sequenced, 358 but sequencing has become so sensitive and efficient that it became possible to sequence and deconvolute complex microbial communities (termed ‘metagenomics’), such as those in soil, sea- and fresh-water, mining and industrial sites, extreme environments, and the digestive tracts of ruminants and humans. 359 Indeed this is the only way to characterize the vast majority of prokaryotic life on earth, which cannot be cultured as (single) colonies on artificial media such as agar plates, 360 although this may be changing, 361 not to mention the estimated billion viruses m in every cubic meter of the ocean. 366
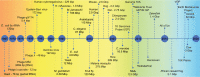
Timeline illustrating the major genome sequencing achievements from the mid-1960s to 2019, placed in a color-coded background according to the sequencing approach. Orange, early sequencing methods; yellow: Sanger-based shotgun sequencing; green: ‘next (more...)
A subset of metagenomics is the human ‘microbiome’ – the bacteria, archaea, protists and fungi, and their own viruses, such as bacteriophages, that inhabit our gut and other places (skin, mouth etc.), and which vastly outnumber our own (human) cells – termed a human “supra-organism”. 367
The human microbiome appears to have a large influence on health, including metabolic activity, autoimmune and inflammatory disorders, atherosclerosis and cancer, 368–374 neurodegenerative and neurodevelopmental disorders, 375–378 neurotransmitter biosynthesis, 379 , 380 social development, 381 , 382 depression, 383 sensory and locomotor behavior, 384 , 385 stress responses, 386 obesity 387 and immunity, 388 all of which are associated with particular types of gut bacteria and bacteriophages. The Human Microbiome Project was initiated in 2007. 367 , 389
A similar exploration of the hugely varied world of protists and fungi is also underway and is reshaping the eukaryotic tree of life. 390 There are far too many projects to catalog here, except to say that it is now or soon will be unacceptable to study any species or ecosystem without sequencing the genomes involved.
And so it is with plants and animals: for example, the 1,000 Plant Genomes Project initiated in 2008 391 – over 200 angiosperm (flowering plant) genomes and over 1000 plant transcriptomes had been sequenced by 2019; 392 , 393 the 10K Vertebrate Genomes Project initiated in 2009; 394 , 395 the ‘Earth BioGenome Project’ initiated in 2018 to characterize the genomes of all of Earth’s eukaryotic biodiversity; 396 the genomes of hundreds of butterfly species; 397 , 398 and the ‘Zoonomia Project’ to characterize the genomes of eutherian mammals, with 131 assemblies reported in 2020. 399 A comparative analysis of 363 bird genomes in 2020 more than doubled the fraction of bases that are predicted to be conserved between species and revealed extensive patterns of selection in non-coding DNA. 400
Despite the numerous (now mainly computational) challenges, genome databases are moving beyond simple gene catalogs to encompass the diversity of variations (nucleotide substitutions, insertions and deletions, as well as structural changes, rearrangements and transposon insertions), and the presence or absence of particular genomic regions in individuals, populations and clades (the ‘pan-genome’ of a species), to allow a greater exploration of genome dynamics and the basis of phenotypic diversity. 38 , 321 , 401–403
The examination and comparison of the evolution and divergence of genomes and their sequence elements 404–407 is an enterprise that will continue for the foreseeable future. Analysis of the genomes of extinct hominids such as Neanderthals and Denisovans by Svante Pääbo and colleagues and others is revealing the details of recent human evolution, 408–412 indicating that there have been multiple bursts of adaptive changes specific to modern humans during the past 600,000 years involving genomic regions related to brain development and function. 413 Others are documenting the diversity in the human population 414 and the details of the migrations out of Africa, 415–420 the provenance of the biblical Dead Sea scrolls, 421 and the genomes of extinct megafauna, such as the mammoth 422 and cave bear. 423
In the years following the completion of the pioneering projects, many studies were described as “genome-wide” or “global”, even though they were limited to protein-coding genes or the ‘exonic’ component of the genome, again on the assumption that most of the relevant information resides therein.
Fortunately, other studies extended to the whole genome, allowing the discovery of many dynamic features outside of coding sequences. The dramatic improvement in sequencing technologies enabled the implementation of unbiased methodologies to globally study the dynamic properties of genomes, including the progressive identification of all transcribed sequences (the ‘transcriptome’) and the positional modifications of histones and DNA (the ‘epigenome’), as well as protein-binding sites, chromatin structure and other features in different cell types at single cell resolution (Chapters 13–14).
Pombe is the Swahili word for beer.
This approach is easier in bacteria because of the low frequency of repetitive sequences whose locations are ambiguous.
Prokaryotic genomes range in size from just 160 kb in the insect symbiotic bacteria Carsonella ruddii 31 and Nasuia deltocephalinicola 32 (and are generally small in endosymbiotic and obligate intracellular parasitic species, such as Mycoplasma) to 14.8 Mb in the free-living soil bacterium Sorangium cellulosum, 33 with ~11,500 protein-coding genes, which appears to be close to the upper limit 34 (Chapter 15).
The standard laboratory strain of E. coli (‘K-12′) has ~4,300 protein-coding genes. 29
At the time of writing, the smallest known eukaryotic genome is that the microsporidian parasite Encephalitozoon intestinalis (2.3 Mb). The largest known plant genome is that of the monocot Paris japonica (~150 Gb). The largest known animal genome is that of the genome of the marbled lung fish, Protopterus aethiopicus (~130 Gb) (Chapter 7). 40
The extent to which alternative splicing expands the proteome may be limited. While it is clear that alternative splicing can increase greatly the number of isoforms of particular proteins, such as in the classic example of the Drosophila Dscam (Down syndrome cell adhesion molecule) gene, which expresses over 38 thousand distinct mRNAs), 109 , 110 much of the alternative splicing in mRNAs, especially in humans, occurs among 5′ non-coding regulatory exons, not within the body of the protein-coding exons, 111 and a large proportion generates non-coding transcripts (Chapter 13). Most protein-coding genes express a single dominant splice isoform. 112
The sequences of 101 Drosophilid genomes have recently been published. 117
Retroviruses and TEs are thought to share an evolutionary relationship. Similar to retroviruses, ERVs and LINEs encode a reverse transcriptase and mobilize via an RNA intermediate. 178
The dark form is long thought to be positively selected because it provided better camouflage from bird predation in a sooty environment (Chapter 2).
Altering the pattern of expression of action of a distal regulatory ‘enhancer’. 314
Open Access: This content is Open Access under the Creative Commons license CC-BY-NC-ND.
Your browsing activity is empty.
Activity recording is turned off.
See more...