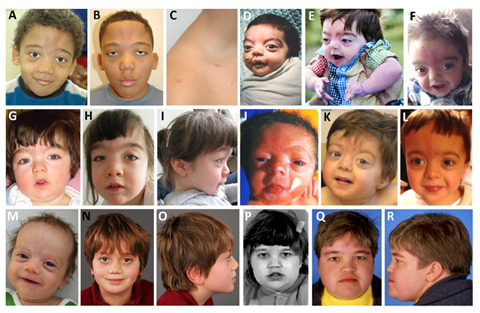
Clinical Description
Shashi-Pena syndrome is characterized by distinctive facial features accompanied by variable further clinical findings, which may include macrocephaly, dental anomalies, congenital heart defects, seizures, hypoglycemia, hypotonia, developmental delays / intellectual disabilities, skeletal abnormalities, and global volume loss on brain MRI.
To date, at least 23 individuals have been identified with pathogenic truncating variants in ASXL2, including published and non-published individuals [Shashi et al 2016; Alqaisi & Hassona 2022; Murphy et al 2022; Ho et al 2023; Yuan et al 2023; Zheng et al 2023; JM Porter, LDM Pena, RC Spillmann, A Johnson, & V Shashi, unpublished data]. The following description of the phenotypic features associated with this condition is based on these individuals.
Table 2.
Shashi-Pena Syndrome: Frequency of Select Features 1, 2
View in own window
| Feature | Number of Persons w/Feature | Comment |
|---|
| Glabellar nevus simplex | 23/23 (100%) | |
| Distinctive facial features | 22/22 (100%) | See Suggestive Findings. |
| Dental abnormalities | 14/14 (100%) | Early eruption & loss; small, fragile teeth |
Developmental delay /
intellectual disability | 20/21 (95%) | Variable, ranging from low-average intellectual abilities to severe intellectual disabilities |
| Hypotonia | 19/20 (95%) | Gradual improvement w/age; likely to be central |
| Other skin findings | 17/20 (85%) | Capillary malformations (11/20), deep palmar creases (6/20), hypertrichosis (4/20) |
| Feeding difficulties (newborn) | 17/20 (85%) | May require nasogastric/gastrostomy tube feeding, but usually resolve over time |
| Feeding difficulties (childhood) | 5/18 (28%) | |
| Skeletal anomalies | 14/17 (82%) | May incl scoliosis/kyphosis, hypermobility, &/or frequent fractures |
| Congenital heart defects | 17/21 (81%) | ASD, PFO, PDA |
| Macrocephaly | 15/20 (75%) | Congenital & acquired both reported |
| Vision abnormalities | 11/15 (73%) | Strabismus, amblyopia; ptosis is part of the distinctive facial features |
| Behavior problems | 10/14 (71%) | Aggression, anger outbursts |
| Seizures | 12/18 (67%) | Febrile &/or non-febrile |
| Hypoglycemia | 13/20 (65%) | Most often in neonates & may be due to hyperinsulinism; frequently resolves over time |
| Macrosomia | 10/18 (55%) | |
| Hearing loss | 7/15 (47%) | Typically conductive |
| Sleep apnea | 7/15 (47%) | May be obstructive or mixed central/obstructive |
| Temperature dysregulation | 6/14 (43%) | |
ASD = atrial septal defect; PFO = patent foramen ovale; PDA = patent ductus arteriosus
- 1.
- 2.
Data points were missing for some individuals, so the denominator for each feature is indicated in the table.
Skin. Glabellar nevus simplex has been seen in all individuals diagnosed with Shashi-Pena syndrome but may fade with age. Other dermatologic findings include deep palmar creases, hypertrichosis, and capillary malformations.
Craniofacial features. Individuals with Shashi-Pena syndrome demonstrate a distinct facial gestalt summarized in Suggestive Findings (see ). Notably, these features tend to be more evident at younger ages and may not be as readily recognizable in older individuals.
Dental. Teeth often erupt early and are small with weak enamel [Alqaisi & Hassona 2022]. Likewise, primary teeth are often lost early with early eruption of secondary (permanent) teeth. Additional dental features include:
Developmental delay (DD) and/or intellectual disability (ID). To date, almost all identified individuals with Shashi-Pena syndrome have demonstrated some degree of developmental delay and/or intellectual disability ranging from mild to severe.
Gross motor delays have manifested as delayed walking (range: age 18 months to after age 4 years).
Verbal communication may be achieved as early as age two years, although some affected individuals continued to be nonverbal at age four years.
The range of intellectual abilities extends from low-average IQ (reported in one such person) to severe ID.
Other neurologic features
Hypotonia is observed in almost all infants with Shashi-Pena syndrome and tends to improve as affected individuals enter childhood to adolescence.
Temperature dysregulation. Several affected individuals reported overheating easily and higher basal body temperatures.
Seizures have been reported in about two thirds of individuals with Shashi-Pena syndrome.
Of these, about one third have febrile seizures only, one third have non-febrile seizures only, and one third have both febrile and non-febrile seizures.
Among non-febrile seizures, generalized tonic-clonic, focal, and absence seizures have been described.
There is no specific pattern of EEG changes noted in people with Shashi-Pena syndrome, but EEG findings tend to be consistent with the reported seizure type.
Seizures are typically well controlled with standard anti-seizure medications when needed (see
Management).
Neurobehavioral/psychiatric manifestations. Anger outbursts and aggression are common and can be difficult to manage. No specific treatment has been found to be superior for behavioral challenges. Anxiety, attention-deficit/hyperactivity disorder, and autism spectrum disorder, either as single diagnoses or in combination, have been observed in a few affected individuals, but there is not enough data to determine if these are consistent findings in individuals with Shashi-Pena syndrome.
Growth. At least two thirds of individuals with Shashi-Pena syndrome have macrosomia, which may be associated with advanced bone age. Macrocephaly is present in about three quarters of affected individuals. Macrocephaly and macrosomia may be present at birth or develop over time.
Gastrointestinal problems. The majority of affected individuals demonstrate feeding difficulties from infancy, most likely due to low tone. Typically this is transient and may be accompanied by poor uncoordinated suck/latch, which can translate into oral apraxia in childhood. Dysphagia and aspiration are not common findings in this condition, and affected individuals are typically able to eat by mouth as children and into adulthood. However, some affected individuals experience sensory issues / food texture aversions.
Musculoskeletal features. Skeletal abnormalities are common in Shashi-Pena syndrome.
About two thirds of affected individuals have scoliosis/kyphosis (11/16; 69%) and/or joint hypermobility (9/14; 64%), although without frank joint dislocations.
Scoliosis/kyphosis may require significant surgical intervention.
A predisposition to fractures (5/17; 29%) and hip dysplasia have been noted in fewer than one third of affected individuals.
Cardiac abnormalities. Cardiac anomalies have been identified in about 80% of people with Shashi-Pena syndrome. The most common reported findings are atrial septal defects or patent ductus arteriosus. Other rare findings may include the following:
Ventricular septal defects
Coarctation of the aorta
Pulmonary artery valve thickening
Pulmonary artery stenosis
Ophthalmologic involvement. About three quarters of people with Shashi-Pena syndrome have ophthalmologic findings, most commonly strabismus and amblyopia. Ptosis is one of the distinctive facial features seen in affected individuals but does not typically impact vision.
Endocrinologic. More than half of affected individuals have hypoglycemia at some point in life.
The exact presentation is variable. Some affected individuals experienced hypoglycemia only in the neonatal period, others had episodic hypoglycemia, and one affected individual had severe hypoglycemia requiring continuous feeds.
Hyperinsulinism has been documented in at least two affected individuals, and one was successfully treated with octreotide [
Yuan et al 2023] (see
Management).
Hearing loss has been identified in approximately half of reported individuals. This is typically conductive hearing loss and may normalize after intervention such as pressure-equalizing (PE) tubes. Multiple sets of PE tubes may be needed. One affected individual had sensorineural hearing loss and received cochlear implants.
Respiratory issues. Nearly half of reported individuals have sleep apnea. While this has not been well characterized in people with Shashi-Pena syndrome, obstructive and mixed obstructive/central sleep apnea has been described. Some affected individuals require positive pressure ventilation during sleep.
Other reported findings