Introduction
This article will review the embryological development of the testicles. However, to understand the embryology of the testicles, it is essential to have a basic understanding of the normal anatomy of the testicles.
The male gonads, otherwise known as the testicles, are sex glands that have both an exocrine secretory function in the production of sperm and an endocrinological function as part of the hypothalamic-pituitary-gonadal axis in men through the production of androgens. The normal anatomy of the testicles is that of an oval shape located in the scrotum, further separated by the scrotal septum. The length of the testis is between 3 cm to 5 cm, whereas the width is between 2 cm to 3 cm.
The consistency of normal testicles on palpation is smooth and soft. The testes are suspended superiorly by the spermatic cord and inferior to the scrotum by the scrotal ligament. During embryological development, the scrotal ligament is also known as the gubernaculum.[1][2]
The tunica vaginalis is a double-layered structure that covers all of the testes apart from the posterior and superior borders, which represent the attachment of the epididymis and spermatic cord. The posterior lateral testis has a small space between the body of the epididymis and the testis. This small space is known as the sinus of the epididymis. The tunica albuginea is found deep in the tunica vaginalis. It is a thick fibrous sheath that covers the testes.
The descent of the testicles is a complex stepwise process that involves an interaction between many anatomical structures, environmental influences, regulatory hormones, and inherited genetic factors.
Development
Development and Sex Determination of Gonads
The beginning of testicular development is with the formation of the genital ridge. The origin of the genital ridge is from the intermediate mesoderm. The intermediate mesoderm forms paired structures that reside beside the dorsal mesentery, specifically in the coelomic cavity.
The proliferation of coelomic epithelial cells found on the ventromedial surface of the mesonephroi initiates the formation of the genital ridge. The mesonephros contains the mesonephric duct (Wolffian duct). This structure is a primordial urogenital tissue that contributes to the formation of the epididymis, seminal vesicles, and vas deferens after male sex determination.[3][4]
The paramesonephric duct (Mullerian duct) is also found in the mesonephros and runs in parallel to the mesonephric duct. This structure is essentially the female equivalent of the mesonephric duct (Wolffian duct) and contributes to the formation of the uterus, fallopian tube, and vagina following female sex determination. The mesonephros and genital ridge form a structure known as the urogenital ridge (UGR).[5]
A dense layer of pseudostratified epithelial cells forms following the proliferation of coelomic epithelial cells, which are present on the ventromedial side of the mesonephros. Basement membrane fragmentation facilitates the migration of epithelial cells dorsally towards the mesonephros.[6] These cells undergo a transition from epithelial type cells to mesenchymal type cells (EMT) and begin to invade the area between coelomic epithelium and mesonephros.
Primordial germ cells (PGC) originate from an area around the yolk sac. PGCs begin to colonize the gonads by approximately five weeks of gestation. By the sixth week of gestation, the PGCs will have invaded the genital ridges. Primordial germ cells divide to reach approximately 3000 cells and undergo further specialization with the activation of gametogenic genes and inactivation of pluripotency genes; this is a process known as licensing.[7]
Following this process of the activation of gametogenic genes and inactivation of pluripotent genes, the primordial germ cells are known as gametogenesis-competent cells, which will either differentiate into male or female specialized cells in response to cues from the cells of the mesonephric tissue.
In genetically male embryos, the primordial germ cells have an XY chromosome complex. In males, there is a sex-specific gene that initiates the determination of gender by activating the production of regulatory sex-determining proteins. This testis-determining factor (TDF) is also known as the sex-determining region of the Y chromosome (SRY). It is the gene resting on the Y chromosome, specifically on its short arm (Yp11).[8] The SRY gene is the only gene on the Y chromosome, which is necessary for testis determination.[9][10] The simple absence of this gene will lead to the development of a female embryo.
The SRY gene encodes for a transcription factor that activates the testis-specific enhancer (TESCO) of a related autosomal gene known as SOX9 (SRY-Box Transcription Factor 9). The SOX9 gene is essential for the initiation of Sertoli cell differentiation from supporting cell precursors. These supporting cells would otherwise differentiate into follicle cells (specifically granuloma cells).[11] Besides SRY, there are other factors such as steroidogenic factor 1 (which is encoded by the gene NR5A1), which contribute to Sertoli cell differentiation.[12] SRY expresses exclusively in the supporting cells in the genital ridges, which differentiate into Sertoli cells.[11]
SRY upregulates levels of SOX9. Once the cellular/tissue levels of SRY and SOX9 are high enough, the SOX9 protein begins to maintain its transcription at high levels in Sertoli cells. Once there are enough SOX9 positive cells in the gonad, it begins to undergo morphological changes, which will lead to the formation of the testes.[13] This process includes the differentiation of interstitial cell lineages (peritubular myoid cells and Leydig cells), the mitotic arrest of germ cells, epithelialization of the Sertoli cells, and the formation of the testicular cords.[14]
By approximately the fifth to the sixth week, the gender of the embryo is determined.
Testicular Descent
There is a biphasic model for the explanation of testicular descent. This process divides into two morphologically and hormonally distinct stages.[15] The first stage of testicular descent (transabdominal) occurs by the eighth gestational week.
Transabdominal Stage
The transabdominal phase of testicular descent consists of the movement of the testes from their starting position on the posterior abdominal wall adjacent to the kidney down to the internal inguinal ring. This phase is dependent on a non-androgenic hormone and starts by the eighth gestational week. Between the eighth and fifteenth gestational weeks, the gubernaculum enlarges causally, leading to the formation of the gubernacular bulb, whereas between the 10th to 15th gestational weeks, the cranial suspensory ligament (CSL) regresses. Regression of the CSL is a necessary step for testicular descent; however, regression of the CSL alone is not sufficient to initiate gonadal descent.[16]
The gubernaculum anchors the testes to the future inguinal region by traction. The gubernaculum shortens as it progressively incorporates into the gubernacular bulb.[17] The enlargement of the gubernaculum during TTD is the "gubernacular swelling reaction" (GSR). Cellular proliferation and extracellular matrix deposition lead to enlargement of the caudal gubernaculum.[18][19] The growth of the gubernaculum is non-androgen dependant. Leydig insulin-like hormone (Insl3) is a "relaxin-like factor" and is a member of the relaxin family of proteins. It contributes to the TTD stage of testicular descent via the activation of the leucine-rich repeat-containing G-protein coupled receptor 8 (LGR8).[20]
Inguinoscrotal Stage
The second stage of testicular descent (inguinoscrotal) begins by the 26th gestational week. This stage is androgen-dependent and consists of the passage of the testes from the internal inguinal ring down into the scrotum.[21] The inguinoscrotal stage of testicular descent (ISTD) has specific pre-requisite steps, includes the formation of the processus vaginalis, dilation of the inguinal canal by the gubernacular bulb, and abdominal pressure to push the testes through the inguinal canal. The processus vaginalis is a herniation of the abdominal peritoneum. It develops as the gubernaculum elongates caudally. When the processus vaginalis enters the caudal abdominal wall, the development of the inguinal canal starts. After testicular descent has finished, the gubernaculum regresses, leaving behind a remnant testo-scrotal attachment. The contraction of the gubernaculum, along with intra-abdominal pressure, facilitates the movement of the testes through the inguinal canal into the scrotum.
The scrotum is effectively a continuation of skin and subcutaneous tissue lining the anterior abdominal wall. As the testes descend through the inguinal canal, it takes layers of the abdominal wall with it. The layers of the abdominal wall involved in testicular descent continue as analogous layers in the scrotum. The processus vaginalis obliterates through a process of programmed smooth muscle cell death in response to a decrease in androgen levels in the third trimester.[22] From superficial to deep, the Scarpa's fascia of the abdominal wall contributes to the dartos fascia and smooth muscle of the scrotum. The fascia of the external oblique muscle contributes to the external spermatic fascia. The internal oblique and transversus abdominis muscles and aponeurosis contribute to the cremasteric fascia and muscle. The transversalis fascia contributes to the internal spermatic fascia. The peritoneum of the abdominal wall contributes to the tunica vaginalis.[23]
In the majority of fetuses, the testes will have descended into the scrotum by the 33rd gestational week.[24] Whereas by the 25th + 4 gestational week, only 7.7% of testes will have descended into the scrotum.[25] By the 27th gestational week, only 12.5% of the testes will have descended into the scrotum.[25] Hence by birth, the testes will be in the scrotum, and it is advisable for the pediatricians to routinely check the scrotum of the newborn to confirm the descent.
Cellular
Sertoli Cells
Sertoli cells are epithelial supportive cells found in the seminiferous tubules. They support the germ cells by providing the required nutrients to complete spermatogenesis.[26] Sertoli cells also undertake a range of other roles, such as contributing to the formation of the blood-testis barrier, phagocytosis of apoptotic spermatocytes, and the secretion of endocrinological signals.[26]
Also known as sustentacular cells, they are the only somatic cells in the entire seminiferous epithelium.[27] They are pyramidal-shaped, with an oval nucleus towards the base. The cells rest on a basement membrane. A large number of germ cells under different stages of development, especially the spermatids, are seen to be embedded near its apex. The spermogonia and spermatocytes invaginate near the base. The base of the Sertoli cells points towards the peritubular myoid cells (PMCs) surrounding the seminiferous tubule, and the apex points towards the lumen of the seminiferous tubule.[26] Inter-Sertolian junctions at the basal lamina of the seminiferous tubules contribute to the formation of the blood-testis barrier. Sertoli cells transport new primary spermatocytes through an active process towards the adluminal compartment of the seminiferous tubules.[28]
Sertoli cells have an epithelial nature. Sertoli cells are the target of hormones such as FSH and testosterone. This activity leads to the regulation of spermatogenesis through regulatory cross-talk between Sertoli cells and germ cells.[29]
Functions of The Sertoli Cells
Not only provide support to the developing germ cells but also house them, provide with required nutrition, and protect them.
[30]Secrete lipids, carbohydrates, vitamins, amino acids, and metal ions required for the proper development of the developing germ cells.
[31]Regulate cholesterol metabolism during spermatogenesis, which plays a vital role in sperm production and male fertility.
[31]Secrete hormone inhibin B, which helps in suppressing the secretion of FSH, thus bettering the function of Sertoli cells, spermatogenic status, and sperm number.
[32][33]Act as macrophages by engulfing the apoptotic bodies, bacteria, all the dead, and worn-out cells.
[34]Secrete an androgen-binding protein (ABP)
[31]Convey the mature germ cells to the lumen to be transported further.
Leydig Cells
The primary source of steroid hormones in a normal male is the Leydig cell. The normal function of the Leydig cell through the production of steroid hormones ensures normal reproductive function and fertility in males.[35] Besides the production of steroid hormones, the Leydig cells have many other essential functions, such as the production of POMC derivatives, calcium-binding proteins, and growth factors.[36]
They are generally large polyhedral, pale cells with an eccentric nucleus lying in the connective tissue present between the seminiferous tubules. The cytoplasm contains rod-shaped Reinke's crystalloid bodies and vacuoles, which are yellowish in color containing various enzymes.[37] Mature adult Leydig cells originate from undifferentiated mesenchymal-like stem cells, which are present in the interstitial part of the early testis.[38]
Molecular Level
As mentioned earlier, there are specific genetic and molecular messengers that are essential for normal testicular descent.
SRY
In 1990 Sinclair et al. identified a gene on the Y chromosome, which they named the sex-determining region of the Y chromosome.[9] This gene is within a 35-kb sex-determining region on the Y chromosome. This gene encodes a 204-amino acid protein which has a molecular mass of 24kD. A DNA binding HMG motif is in the middle of this protein.[39] It is a member of the high mobility group (HMG)-box of DNA binding proteins.
The SRY gene becomes transactivated by the product of the WT1, which binds to the SRY promoter region.[40] SRY binds to a testis-specific enhancer of SOX9 core (TESCO) in mice along with steroidogenic factor 1 (SF1).[12] SF1 and SRY work to upregulate levels of SOX9. When the production of SRY reduces SOX9, and SF1 maintains its expression.
SOX9
The SOX9 (SRY-Box Transcription Factor 9) gene encodes a 509 amino acid long transcription factor, which is necessary for sexual development. It is found on the long arm of chromosome 17 and is a member of HMG transcription factors having a molecular mass of 56kD.[41][42]The SRY gene initiates a sequence of gene interactions along with SOX9, which is necessary for testicular development.[12]
SRY begins a feed-forward loop between Glia-activating factor (FGF9) and SOX9; this causes the upregulation of FGF9 and inhibits WNT4. WNT4 promotes female sex development, whereas FGF9 stimulates male sex development. This upregulation of FGF9 and SOX9 and downregulation of WNT4 stimulate testis development.[43] In conjunction with steroidogenic factor 1, SOX9 can stimulate the production of anti-mullerian hormone (AMH) to inhibit female sexual differentiation.[44]
SF1
Steroidogenic factor 1 (SF1), also known as NR5A1, is a transcription factor that consists of 461 amino acids.[45] SF1 is present in primary steroidogenic tissues, including the adrenal cortex, ovarian theca cells, and Leydig cells of the testis. Along with SOX9, SF1 regulates the production of AMH.[46][44]
AMH
Anti-Mullerian hormone (AMH), also known as Mullerian inhibiting substance (MIS) is a 560 amino acid long protein.[47] The gene for AMH is on chromosome 19.[48] AMH functions to prevent the development of the Mullerian ducts into female structures.[49] The suppression of the development of Mullerian structures takes place within the first eight weeks of development. AMH is also produced in motor neurons and is suspected to be responsible for subtle behavioral differences between males and females.[50][51]
INSL3
Insulin-like peptide 3 (INSL3), otherwise known as Leydig cell-specific relaxin-like factor, is a hormone encoded by the INSL3 gene found on chromosome 19.[52] It has a similar structure to insulin and relaxin and belongs to the “insulin-like hormone superfamily,” which includes other hormones such as insulin, relaxin, and insulin-like growth factors (I and II).[53] INSL3 is essential for the development of the gubernaculum in male embryos and is a crucial factor in testicular descent.[54]
Function
The Normal Function of the Testes
There are two main functions of the testes. These are the endocrine production of androgens and exocrine production of spermatozoa.
The endocrine function of the testis is under neuroendocrine control. The production of gonadotrophin-releasing hormone (GnRH) by the anterior hypothalamus stimulates the production of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in the anterior pituitary.[55] LH and FSH bind to receptors found on Sertoli and Leydig cells. Luteinizing hormone stimulates Leydig cells to produce testosterone, whereas FSH stimulates Sertoli cells to produce androgen binding proteins (ABPs) along with the regulation of spermatogenesis.[56]
Endocrine Function
GnRH is released in repeated pulses in response to excitatory neurotransmitters such as glutamate and kisspeptin.[57][58] GnRH binds to a G-protein seven-transmembrane receptor (GPCR) known as the gonadotrophin-releasing hormone receptor (GnRHR) on gonadotrophic cells of the anterior pituitary. FSH becomes stimulated when GnRH pulses are a low frequency, whereas stimulation of LH occurs when GnRH pulses are at a high frequency.[59][60]
Luteinizing hormone stimulates the production of androgens in Leydig cells by binding to a GPCR.[61] During testicular development, blood testosterone levels peak three times. The first time is between 12 to 14 weeks of gestation when Leydig cell development occurs.[62] The second time is two months post-partum, which leads to the proliferation of Leydig cells. However, there is subsequent atrophy of Leydig cells leading to the interstitium of the testis becoming inhabited by inactive precursor cells. Mature adult Leydig cells develop during puberty.[63] The third peak of testosterone is during the onset of puberty.
Leydig cell androgen production is dependent on the activity of four enzymes. These are cytochrome P450 17a-hydroxylase (P450 17a), P450 side-chain cleavage enzyme (P450scc), 17b-hydroxysteroid dehydrogenase (17b HSD) and 3b-hydroxysteroid dehydrogenase (3b-HSD).[64] Leydig cells convert cholesterol into pregnenolone, and pregnenolone then converts into progesterone. Progesterone then metabolizes into testosterone. A mutation in the genes that code for the enzymes P450 17a, P450scc, or 3b-HSD can cause congenital adrenal hyperplasia, whereas a mutation in the gene for 17b-HSD causes 17b-HSD deficiency. These can all present with impaired virilization.
Exocrine Function
Spermatozoa are produced from spermatogonia through a stepwise process. The spermatogonia are at the basement membrane of the seminiferous tubules. The spermatogonia can be either stem cells (type A spermatogonia) or differentiated spermatogonia (Type B spermatogonia). Type A spermatogonia either produce more type A spermatogonia or type B spermatogonia.[65][66][67] Type B spermatogonia divide and produce primary spermatocytes. The primary spermatocytes then enter the prophase of meiosis 1. During this period, they move away from the basement membrane towards the lumen of the seminiferous tubule.
After completing the prophase of meiosis 1, the spermatocytes rapidly complete meiosis 1. This process results in the formation of haploid secondary spermatocytes. The secondary spermatocytes complete meiosis 2, leading to the production of four haploid spermatids from one primary spermatocyte.[55] Spermatids undergo spermiogenesis, the process during which the spermatids become spermatozoa and acquire the characteristic features of spermatozoa, such as the formation of a tail and loss of cytoplasm.[55] Sertoli cells, also known as ‘nursemaid cells,’ provide nutrients, growth factors, and organization to the developing spermatids.[68]
Functional Significance of Testicular Descent
According to the ‘temperature-dependent hypothesis,’ testicular descent is necessary because a low-temperature environment is needed to produce viable sperm. Normal spermatogenesis and storage in the epididymis require temperatures below that of the core body temperature.[69][70][71]
A testicular temperature of 2 to 7 degrees C below core body temperature is a requirement for effective spermatogenesis.[72][69] The testes have anatomical features that optimize temperature regulation. Thin scrotal skin with a large number of sweat glands, the pampiniform plexus which is a venous plexus surrounding the testes, the cremaster muscle which will contract or relax in response to temperature, the tunica dartos, and a lack of adipose tissue also have an essential role in regulating the temperature.[69]
Testing
Imaging
In the context of undescended testis, imaging methods can serve to determine the anatomical location of undescended testes; this allows for better pre-surgical planning and reduces the extent of exploration and anesthesia time.[73] Magnetic resonance imagining (MRI) and ultrasound (US) are useful in diagnosing and locating undescended testis. These techniques are preferable to others as they are non-invasive and do not use ionizing radiation. The diagnostic accuracy of MRI and US are similar to other methods.[74][75][74]
Karyotyping
Chromosomal changes are detectable through the use of a technique known as karyotyping, which allows the identification of the chromosomes so that in situations where there may be an extra or missing chromosome, it is detectable through karyotyping.[76] Karyotyping is a recommended investigation in all patients who present with disorders of sexual differentiation such as cryptorchidism and proximal hypospadias.[77]
Pathophysiology
Testicular Developmental Disorders
Testicular Dysgenesis
Testicular dysgenesis syndrome (TDS) is a constellation of poor-quality semen, testicular germ cell cancer, cryptorchidism, and hypospadias. It is a consequence of disruption to embryonal programming and gonad development.[78] The belief is that testicular dysgenesis syndrome is the result of environmental disrupting chemicals (EDCs), intrauterine growth disorders, genetic and lifestyle factors.
Reduced production of androgen receptors, disrupted hormone levels along with exposure to endocrine disrupters are associated with the development of TDS.[79] The thought is that this occurs due to Leydig-cell dysfunction and reduced INSL3 secretion from Leydig cells, which consequently leads to the disorders associated with TDS.[80] Furthermore, a disruption in Sertoli cell development contributes to the development of TDS.[81]
As mentioned before, researchers believe that EDCs contribute to the development of TDS through the interference with the production, transport, metabolism, binding, release, action, and elimination of hormones associated with developmental processes.[82] This result can be through anti-androgenic effects, estrogenic effects of steroidogenic enzyme inhibitory effects.[83][84]
There are historical examples of maternal exposure to chemicals contributing the testicular developmental disorders; the synthetic estrogenic drug known as diethylstilboestrol (DES) was historically used for the prevention of the complications of pregnancy but was shown to increase the risk of testicular malformations.[85] Another environmental toxin that appears to contribute to testicular malformations is bisphenol A (BPA). This chemical has weak estrogenic effects and is used in the production of polycarbonates and epoxy resins.[86] Phthalate esters are used in vinyl plastics to improve their flexibility. However, it seems that they have an endocrine-disrupting effect.[79]
Cryptorchidism
It is a state wherein there is an absence of the testes in the scrotum may be unilaterally or bilaterally. The etiology of cryptorchidism can divide into anatomical, genetic, or hormonal causes.[87][88] Anatomical causes of cryptorchidism include conditions that cause an anatomical abnormality of the inguinal canal, vas deferens, or testis themselves.[89] Genetic causes of cryptorchidism include 5α-reductase gene mutations, androgen receptor mutations, mutations in INSL3 genes.[90][91][90][92] Hormonal causes of cryptorchidism include a deficiency or insensitivity to AMH, INSL3, androgen, or GNRH/LH.[87][93][94] Cryptorchidism is problematic as it can lead to an increased likelihood of infertility and an increased likelihood of developing testicular cancer.[95][93]
As the main functional significance of the descent of the testes according to the ‘temperature-dependent hypothesis’ is the maintenance of a low-temperature environment to produce viable sperm, failure of testicular descent will lead to an increase in the testicular temperature. An increase in testicular temperature causes a reduction in effective spermatogenesis. Furthermore, damage to Sertoli cells may occur due to the production of reactive oxygen species.[69][96]
Cryptorchidism appears to demonstrate associations with an increased risk of testicular intraepithelial neoplasm (TIN), which may lead to testicular carcinoma in situ (CIS).[97] It is thought that the high temperatures associated with cryptorchidism lead to a failure of the development of neonatal gonocytes to differentiate into primary spermatogonia, which allows some gonocytes to persist as a potential source of malignancy.[95][93]
Anorchia
By definition, anorchia is the absence of testes in a person who has a 46 XY karyotype and an external male phenotype. It is also known as vanishing testis syndrome or embryonic testicular regression.[98][99][100] The exact etiology of congenital anorchia is not well known. However, there appears to be a genetic component due to the familial inheritance associated with anorchia.[98] However, in previous studies, mutations in genes such as SRY, INSL3/INSL3 receptor do not have an association with anorchia.[101] But mutations in genes such as NR5A1, which codes for SF1, correlate with anorchia.[102]
Because patients with anorchia have a normal male phenotype, scientists suspect that the testes were functionally normal during some point of embryological development.[103] Other suspected etiologies are testicular torsion, vascular occlusion, and trauma during the descent of the testes.[104]
Monorchism
Monorchism is, by definition, the state of only having one testis in the scrotum. Monorchism may result from trauma, testicular torsion, or surgical removal because of testicular malignancy.[105] Congenital monorchism is when a child is born with only one testis in the scrotum; this can be due to cryptorchidism or vascular pathology, which causes unilateral regression of the testis (anorchia/vanishing testis syndrome) such as spermatic cord torsion or thrombosis.[106][107] Monorchism may also occur in disorders of sexual development (DSD).[108] However, endocrine abnormalities are unlikely to be the sole cause of unilateral congenital monorchism. Men with congenital monorchism do not have a significant difference in hormonal excretory function compared to men with two testes.[109]
Polyorchidism
Polyorchidism refers to a congenital anomaly in which more than two testes are present in the scrotum.[110] Even though this is a very rare condition; 40 % of polyorchidism is associated with undescended testes.[111] The most common type of polyorchidism is triorchidism, which refers to the presence of three testicles.[112] It appears that polyorchidism occurs due to abnormal separation of the genital ridge during the development of the testes in the embryo.[113]
Leung described a way of classifying polyorchidism based on embryological differentiation.[114]
Type I: An extra testis without an epididymis or vas which attaches to the ipsilateral testis.
Type II: The extra testis drains through the epididymis of the ipsilateral testis, and they share a common vas deferens.
Type III: The extra testis has its own epididymis separate from the ipsilateral testis, both of those structures draining into a common vas deferens.
Type IV: The extra testis has a separate epididymis and vas deferens from the ipsilateral testis.
Testicular Torsion
Testicular torsion is a state in which the spermatic cord twisted upon itself, leading to a restriction of blood supply and venous drainage of the ipsilateral testis. If emergency treatment is not delivered quickly, it may lead to necrosis of the testis.
Researchers believe that the majority of postnatal testicular torsion is associated with the "bell clapper anomaly."[115] Hypermobility of the testicle is thought to be inherited through an X-linked dominant or Y-linked mechanism.[116][117][118] The bell clapper anomaly refers to the posterior attachment of the parietal lamina of the tunica vaginalis (TVPL). In a normal testis, the TVPL is attached to the posterolateral aspect of the testis, whereas in patients with the bell clapper anomaly, the TVPL attachment encircles and connects proximally to the spermatic cord, which leads to free-hanging testis in the tunica vaginalis.[119]
Testicular torsion is extravaginal or intravaginal.[120] Extravaginal testicular torsion occurs more frequently in neonates, it is due to the testis, epididymis, and tunica vaginalis twisting along with the spermatic cord.[121] In contrast, intravaginal testicular torsion occurs secondary to the bell clapper deformity.[120]
Swyer Syndrome
Swyer syndrome, also known as pure gonadal dysgenesis, occurs when there is abnormal sexual differentiation during embryological development, leading to insufficient intrauterine virilization with undifferentiated gonads with associated female phenotype.[122] The external genitalia is non-ambiguous and well-formed; however, the gonads are dysgenic and improperly formed.[123] Patients with Swyer syndrome have an XY 46 karyotype.
Swyer syndrome occurs due to reduced production of testosterone and AMH leading to a failure of gonadal differentiation.[122] Particular mutations associated with Swyer syndrome include mutations in NR5A1 (which encodes for SF1), WNT4, and the SRY gene, which mutates in 10 to 15% of patients with Swyer syndrome.[124][123]
Klinefelters Syndrome
Klinefelters syndrome is a common congenital chromosomal disorder with an incidence of 0.1% to 0.2% of newborns.[125] It is a common cause of primary hypogonadism.[126] The majority of patients (80%) have the 47 XXY karyotype, whereas 20% of patients with Klinefelter have supernumerary X chromosomes (49 XXXXY or 48 XXXY), mosaicism of 47 XXY/46XY, or more than one Y chromosome (XXYY).[126][127][128]
Androgen Insensitivity Syndrome
A child can have a disorder of sexual development (DSD) without a chromosomal abnormality such as Klinefelters syndrome. The most commonly seen 46 XY DSD is androgen insensitivity syndrome (AIS).[129] The severity of AIS can range from complete androgen insensitivity syndrome (CAIS), in which female genitalia is present, to mild androgen insensitivity syndrome (MAIS) in which normal external male genitalia is present with or without gynecomastia.[130][131]
AIS occurs due to molecular defects in the gene that codes for the androgen receptor (AR).[132] The gene for the AR is on the X chromosome at position Xq11-12.[133] Mutations in the gene that provides information for the AR known as NR3C4 cause AIS.[134] The AR is a nuclear receptor that binds to androgens such as testosterone and dihydrotestosterone (DHT).
As patients with AIS have a Y chromosome, they will produce male gonads. However, as the fetal cells do not respond to androgens, there will be abnormal development of the reproductive system. This situation may manifest as undescended testes. Furthermore, as androgens play a part in the inhibition of female characteristics through SOX9 and SF1, a loss of response to androgens may lead to a female phenotype.[135]
5-ARD Deficiency
The enzyme 5-alpha reductase 2 (5alpha-RD2) converts testosterone into DHT.[136] As mentioned earlier, androgens are essential during the development of male genitalia. A failure of the conversion of testosterone into DHT will lead to abnormal development of the genitalia.
5-alpha reductase deficiency is a 46 XY DSD. It presents with a female phenotype or improperly virilized external genitalia along with testes. The gene for 5 alpha-RD2, also known as SRD5A2, is found on the second chromosome on position 2p23, it is an autosomal recessive condition.[137][136]
Congenital Adrenal Hyperplasia
The most common inborn error of adrenal function is congenital adrenal hyperplasia (CAH) secondary to a deficiency of the steroid 21-hydroxylase enzyme.[138] This condition occurs due to deletions or mutations in the gene CYP21A2, which codes for the steroid 21-hydroxylase enzyme found on chromosome 6p21.[139][140] This mutation leads to an impairment of the production of cortisol and aldosterone alongside excessive adrenal androgen production. However, this form of congenital adrenal hyperplasia does not cause anatomical disorders in males.
In infant females, the most common form of congenital adrenal hyperplasia (21-hydroxylase deficiency) may present with a salt-wasting disease or virilization of the external genitalia. In both males and females, it can present with precocious puberty.[140]
Other less common forms of congenital adrenal hyperplasia include the 3b-hydroxysteroid dehydrogenase (3b-HSD) deficiency which occurs due to mutations in the HSD3B2 gene, the 17a-hydroxylase (P450 17a) deficiency which occurs due to mutations in the CYP17A1 gene, and the P450 side-chain cleavage enzyme (P450scc) deficiency which occurs due to mutations in the CYP11A1 gene.[140]
A deficiency in the 3b-HSD enzyme causes a failure of the conversion of 17-a hydroxypregnenolone to 17a hydroxyprogesterone, pregnenolone to progesterone, and DHEA to androstenedione, resulting in the accumulation of pregnenolone, DHEA, and 17-a hydroxypregnenolone and a reduction in the production of sex steroids. It may also cause salt-wasting disease.[141] Reduced production of androgenic steroid hormones may present as pseudohermaphroditism associated with cryptorchidism.[141]
A deficiency in the P450 17a enzyme leads to a failure of the conversion of pregnenolone to 17a-hydroxypregnenolone, progesterone to 17a- hydroxyprogesterone, and reduced conversion of 17-hydroxypregnenolone into DHEA. This process leads to the accumulation of pregnenolone, progesterone, and reduced production of glucocorticoids and sex steroids, leading to incomplete virilization in males associated with ambiguous external genitalia and cryptorchidism.[142][143]
A deficiency in the P450scc enzyme leads to a failure of the conversion of cholesterol into pregnenolone; this is the first step in the synthesis of all steroid hormones. As a result, there is a severe deficiency in steroid hormones.[144] Regardless of the chromosomal gender, children with a mutation in P450scc are born phenotypically female.[145] However, due to a failure of the production of androgens, the testes fail to descend.[146]
Other Related Conditions
Congenital Inguinal Hernia
The lifetime risk of inguinal hernias is significantly higher in men (27%) than in women (3%).[147] As such, one of the main risk factors for a primary inguinal hernia is the male gender.[148] Other risk factors for primary inguinal hernias include patency of the processus vaginalis. Obliteration of the processus vaginalis occurs typically by the third trimester through programmed cell death of smooth muscle cells.[149] Non-obliteration of this process vaginalis results in abnormal protruding of abdominal contents through the inguinal canal into the scrotum.
Clinical Significance
Testicular Developmental Disorders
Cryptorchidism
Congenital undescended testes (cryptorchidism) is a very common congenital urogenital malformation with a prevalence among normal birth weight boys varying between 1.8 to 8.4%.[150]
By definition, cryptorchidism is a condition where either one or both of the testes do not descend into the scrotum.[151] The undescended testes can be found in the usual route of descent or an abnormal ectopic position, leading to a way of classifying undescended testes according to their position:
Cryptorchid – This refers to a testis that is not in the scrotum and cannot be manipulated so that they enter the scrotum.
Ectopic – This refers to a testis that has not followed the regular developmental route of testicular descent. This condition can be femoral, pubopenile, crossed scrotal, or perineal.
[152]Gliding – This refers to a testis that can be manipulated to enter into the upper scrotum, but with the removal of manipulation, they retract.
[153]Retractile – This refers to the testis, which can be manipulated to enter into the scrotum; they do not retract when tension is released.
Acquired – This refers to the testis, which initially had normal descent that ascends spontaneously.
[154]
The diagnosis of cryptorchidism is by manual palpation; this is an essential part of the newborn physical examination and should be diagnosed during infancy.[155] Further investigation of undescended testicles includes various imaging techniques such as ultrasound, CT, and MRI.[73][156]
Anorchia
Anorchia occurs in 1/20,000 males and is also associated with 1/177 of those with cryptorchidism.[157][158] The majority of patients with anorchia have a normal phenotype, but some may present with abnormalities of the external genitalia.[159] The characteristic phenotype in a patient with anorchia is the absence of gonads despite having an XY karyotype. Most cases are associated with small tubular rudimentary Mullerian/Wolffian remnants. The phenotypical presentation can range from phenotypic females with a small, well-formed uterus to a phenotypical male with no testes.[160]
It is crucial to distinguish anorchia from cryptorchidism by examination of the abdomen and inguinal canal. Furthermore, as congenital adrenal hyperplasia can present similarly, plasma 17a-hydroxylase levels should be determined to rule out CAH. The diagnosis of anorchia can be made through undetectable plasma levels of AMH and inhibin B associated with raised plasma levels of FSH.[98] Testosterone therapy can be given to ensure the patient reaches a preferable height during puberty.
Monorchism
Clinically, it is not always possible to determine the exact etiology of monorchism as a child presenting with monorchism may have cryptorchidism, testicular agenesis, or VTS. Further investigation, such as laparoscopy or imaging techniques, may be performed. However, the accuracy of imaging techniques and the invasive nature of diagnostic laparoscopy means are not optimal methods for determining the cause of monorchism.[161] Alternative methods to determine the cause of monorchism is the volume of the contralateral testis in adults. However, in younger patients, this is not useful in predicting the cause of monorchism.[162][161] Fixation of the contralateral testicle is considered in children with monorchism to reduce the risk of torsion later in life to avoid a total loss of reproductive and endocrine function.[105]
Polyorchidism
The majority of patients with polyorchidism are asymptomatic.[163] It is usually a coincidental finding on the investigation of other symptoms such as hernia (30%), torsion (15%), mal-descent (40%), malignancy (6%), and hydrocele (9%).[164][165][166] In the majority of cases, the supernumerary testis is found on the left side [167]. The supernumerary testis may be found in the inguinal region (23%), scrotum (66%), or the abdomen (9%).[164][165]
Testicular Torsion
The prevalence of testicular torsion in males below the age of 25 years is between 2.9 to 3.8 patients per 100,000 people.[168][169][170] The classical clinical presentation of testicular torsion is acute severe scrotal pain, which occurs at rest, associated with some type of trauma. The severe pain can cause the patient to become nauseous or to vomit.[171] Testicular torsion tends to be associated with an absent cremasteric reflex.[171] Testicular torsion has a significant impact on the patient's future fertility and can be associated with psychological trauma.[172] Surgical intervention in testicular torsion has to be prompt. There is a 90 to 100% salvage rate if operation occurs within 6 hours of the onset of testicular torsion. The salvage rate reduces with time.[173]
Testicular torsion is diagnosable using a color doppler ultrasound; this is the gold standard for the evaluation of the acute scrotum.[174] Testicular torsion management involves emergency surgical exploration. Following this, the testis is untwisted and wrapped in a warm soaked gauze. The testis is then observed to see if there is an improvement in the color of the testis. To reduce the likelihood of the contralateral testis undergoing torsion, it is fixed along with the testis, which underwent torsion.[171]
Swyer Syndrome
Swyer syndrome has an incidence of 1 in 80,0000.[122] Swyer syndrome patients present with an above average height, underdeveloped breasts with normal axillary/pubic hair. The external genitalia is distinctly female; however, the uterus is small. The underdeveloped gonads are dysgenetic strips that do not have any endocrinological or reproductive function.[122] Patients with Swyer syndrome have an increased incidence of gonadal tumors such as gonadoblastomas or dysgerminomas.[122] One-third of patients with Swyer syndrome may suffer from dysgerminomas.[122]
Klinefelters Syndrome
In Klinefelter's syndrome, the primordial germ cells fail to develop appropriately such that by puberty, no germ cells remain, resulting in insufficient spermatogenesis.[175] Klinefelters syndrome is also associated with characteristic phenotypical features such as [125]:
5-ARD Deficiency
The phenotypical presentation of 5-ARD deficiency does not tend to match the genotypical presentation such that patients with the same genotype may present with different phenotypes.[176] However, phenotypical presentations include ambiguous genitalia with unfused labioscrotal folds resembling the labia majora or a phallus with a clitoris-like appearance.[177] Nevertheless, the internal genitalia such as the vas deferens, epididymis will be present. Undescended testes are also associated with 5-ARD deficiency.[177]
Congenital Adrenal Hyperplasia
A deficiency in 3B-HSD accounts for between 1% and 10% of CAH cases.[178][179] A deficiency in the P450 17a enzyme accounts for approximately 1% of CAH cases.[180] Deficiencies in the P50scc enzyme are incredibly rare and tend to present with a significant clinical variation.[146]
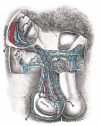
Testicle Illustration. Testicle illustration includes vas (ductus) deferens, head of epididymis (shown lifted from testis), body of epididymis, tail of epididymis, testicular artery, pampiniform plexus, efferent ductules, septa, lobules, and tunica albuginea. (more...)
The Male genital Organs, Spermatic veins, Testis Henry Vandyke Carter, Public Domain, via Wikimedia Commons
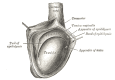
The Male Genital Organs, The right testis, exposed by laying open the tunica vaginalis, Testis, Tail of Epididymis, Cremaster, Tunica Vaginalis Henry Vandyke Carter, Public Domain, via Wikimedia Commons
The Male Genital organs, Transverse section of a tubule of the testis of a rat Henry Vandyke Carter, Public Domain, via Wikimedia Commons
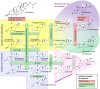
Adrenal Pathway, Mineralocorticoids, Progestagens, Androgens Contributed by Häggström M, Richfield D
References
- 1.
Tsili AC, Sofikitis N, Stiliara E, Argyropoulou MI. MRI of testicular malignancies.
Abdom Radiol (NY). 2019 Mar;44(3):1070-1082. [
PubMed: 30386879]
- 2.
Mäkelä JA, Koskenniemi JJ, Virtanen HE, Toppari J. Testis Development.
Endocr Rev. 2019 Aug 01;40(4):857-905. [
PubMed: 30590466]
- 3.
Yang Y, Workman S, Wilson M. The molecular pathways underlying early gonadal development.
J Mol Endocrinol. 2018 Jul 24; [
PubMed: 30042122]
- 4.
Avellar MCW, Ribeiro CM, Dias-da-Silva MR, Silva EJR. In search of new paradigms for epididymal health and disease: innate immunity, inflammatory mediators, and steroid hormones.
Andrology. 2019 Sep;7(5):690-702. [
PubMed: 31207127]
- 5.
Acién P. Embryological observations on the female genital tract.
Hum Reprod. 1992 Apr;7(4):437-45. [
PubMed: 1522183]
- 6.
Gropp A, Ohno S. The presence of a common embryonic blastema for ovarian and testicular parenchymal (follicular, interstitial and tubular) cells in cattle Bos taurus.
Z Zellforsch Mikrosk Anat. 1966;74(4):505-28. [
PubMed: 5988471]
- 7.
Gill ME, Hu YC, Lin Y, Page DC. Licensing of gametogenesis, dependent on RNA binding protein DAZL, as a gateway to sexual differentiation of fetal germ cells.
Proc Natl Acad Sci U S A. 2011 May 03;108(18):7443-8. [
PMC free article: PMC3088606] [
PubMed: 21504946]
- 8.
Hoare BS, Khan YS.
StatPearls [Internet]. StatPearls Publishing; Treasure Island (FL): Jul 24, 2023. Anatomy, Abdomen and Pelvis: Female Internal Genitals. [
PubMed: 32119488]
- 9.
Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R, Goodfellow PN. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif.
Nature. 1990 Jul 19;346(6281):240-4. [
PubMed: 1695712]
- 10.
Gubbay J, Collignon J, Koopman P, Capel B, Economou A, Münsterberg A, Vivian N, Goodfellow P, Lovell-Badge R. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes.
Nature. 1990 Jul 19;346(6281):245-50. [
PubMed: 2374589]
- 11.
Albrecht KH, Eicher EM. Evidence that Sry is expressed in pre-Sertoli cells and Sertoli and granulosa cells have a common precursor.
Dev Biol. 2001 Dec 01;240(1):92-107. [
PubMed: 11784049]
- 12.
Sekido R, Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer.
Nature. 2008 Jun 12;453(7197):930-4. [
PubMed: 18454134]
- 13.
Warr N, Greenfield A. The molecular and cellular basis of gonadal sex reversal in mice and humans.
Wiley Interdiscip Rev Dev Biol. 2012 Jul-Aug;1(4):559-77. [
PMC free article: PMC3709125] [
PubMed: 23801533]
- 14.
Larney C, Bailey TL, Koopman P. Switching on sex: transcriptional regulation of the testis-determining gene Sry.
Development. 2014 Jun;141(11):2195-205. [
PMC free article: PMC4034426] [
PubMed: 24866114]
- 15.
Hutson JM. A biphasic model for the hormonal control of testicular descent.
Lancet. 1985 Aug 24;2(8452):419-21. [
PubMed: 2863447]
- 16.
Emmen JM, McLuskey A, Grootegoed JA, Brinkmann AO. Androgen action during male sex differentiation includes suppression of cranial suspensory ligament development.
Hum Reprod. 1998 May;13(5):1272-80. [
PubMed: 9647559]
- 17.
Wensing CJ. Testicular descent in some domestic mammals. 3. Search for the factors that regulate the gubernacular reaction.
Proc K Ned Akad Wet C. 1973;76(2):196-202. [
PubMed: 4267621]
- 18.
Heyns CF, Human HJ, De Klerk DP. Hyperplasia and hypertrophy of the gubernaculum during testicular descent in the fetus.
J Urol. 1986 May;135(5):1043-7. [
PubMed: 2421019]
- 19.
Heyns CF, Human HJ, Werely CJ, De Klerk DP. The glycosaminoglycans of the gubernaculum during testicular descent in the fetus.
J Urol. 1990 Mar;143(3):612-7. [
PubMed: 2106043]
- 20.
Kumagai J, Hsu SY, Matsumi H, Roh JS, Fu P, Wade JD, Bathgate RA, Hsueh AJ. INSL3/Leydig insulin-like peptide activates the LGR8 receptor important in testis descent.
J Biol Chem. 2002 Aug 30;277(35):31283-6. [
PubMed: 12114498]
- 21.
Heyns CF. The gubernaculum during testicular descent in the human fetus.
J Anat. 1987 Aug;153:93-112. [
PMC free article: PMC1261785] [
PubMed: 2892824]
- 22.
Tanyel FC. Obliteration of processus vaginalis: aberrations in the regulatory mechanism result in an inguinal hernia, hydrocele or undescended testis.
Turk J Pediatr. 2004;46 Suppl:18-27. [
PubMed: 15499794]
- 23.
Patel AP. Anatomy and physiology of chronic scrotal pain.
Transl Androl Urol. 2017 May;6(Suppl 1):S51-S56. [
PMC free article: PMC5503924] [
PubMed: 28725619]
- 24.
Malas MA, Sulak O, Oztürk A. The growth of the testes during the fetal period.
BJU Int. 1999 Oct;84(6):689-92. [
PubMed: 10510117]
- 25.
Nemec SF, Nemec U, Weber M, Kasprian G, Brugger PC, Krestan CR, Rotmensch S, Rimoin DL, Graham JM, Prayer D. Male sexual development in utero: testicular descent on prenatal magnetic resonance imaging.
Ultrasound Obstet Gynecol. 2011 Dec;38(6):688-94. [
PubMed: 21337441]
- 26.
Barrionuevo F, Burgos M, Jiménez R. Origin and function of embryonic Sertoli cells.
Biomol Concepts. 2011 Dec 01;2(6):537-47. [
PubMed: 25962053]
- 27.
Chojnacka K, Zarzycka M, Mruk DD. Biology of the Sertoli Cell in the Fetal, Pubertal, and Adult Mammalian Testis.
Results Probl Cell Differ. 2016;58:225-51. [
PubMed: 27300181]
- 28.
Cheng CY, Mruk DD. Cell junction dynamics in the testis: Sertoli-germ cell interactions and male contraceptive development.
Physiol Rev. 2002 Oct;82(4):825-74. [
PubMed: 12270945]
- 29.
Silva FR, Leite LD, Wassermann GF. Rapid signal transduction in Sertoli cells.
Eur J Endocrinol. 2002 Sep;147(3):425-33. [
PubMed: 12213681]
- 30.
Ni FD, Hao SL, Yang WX. Multiple signaling pathways in Sertoli cells: recent findings in spermatogenesis.
Cell Death Dis. 2019 Jul 17;10(8):541. [
PMC free article: PMC6637205] [
PubMed: 31316051]
- 31.
Shi JF, Li YK, Ren K, Xie YJ, Yin WD, Mo ZC. Characterization of cholesterol metabolism in Sertoli cells and spermatogenesis (Review).
Mol Med Rep. 2018 Jan;17(1):705-713. [
PMC free article: PMC5780145] [
PubMed: 29115523]
- 32.
Makanji Y, Zhu J, Mishra R, Holmquist C, Wong WP, Schwartz NB, Mayo KE, Woodruff TK. Inhibin at 90: from discovery to clinical application, a historical review.
Endocr Rev. 2014 Oct;35(5):747-94. [
PMC free article: PMC4167436] [
PubMed: 25051334]
- 33.
O'Connor AE, De Kretser DM. Inhibins in normal male physiology.
Semin Reprod Med. 2004 Aug;22(3):177-85. [
PubMed: 15319820]
- 34.
Shiratsuchi A, Osada Y, Nakanishi Y. Differences in the mode of phagocytosis of bacteria between macrophages and testicular Sertoli cells.
Drug Discov Ther. 2013 Apr;7(2):73-7. [
PubMed: 23715505]
- 35.
Svechnikov K, Landreh L, Weisser J, Izzo G, Colón E, Svechnikova I, Söder O. Origin, development and regulation of human Leydig cells.
Horm Res Paediatr. 2010;73(2):93-101. [
PubMed: 20190545]
- 36.
Davidoff MS, Schulze W, Middendorff R, Holstein AF. The Leydig cell of the human testis--a new member of the diffuse neuroendocrine system.
Cell Tissue Res. 1993 Mar;271(3):429-39. [
PubMed: 8472301]
- 37.
Mahran AM, Elgamal DA, Ghafeer HH, Abdel-Maksoud SA, Farrag AA. Histological alterations in Leydig cells and macrophages in azoospermic men.
Andrologia. 2017 Oct;49(8) [
PubMed: 27709649]
- 38.
Chen H, Wang Y, Ge R, Zirkin BR. Leydig cell stem cells: Identification, proliferation and differentiation.
Mol Cell Endocrinol. 2017 Apr 15;445:65-73. [
PMC free article: PMC5346484] [
PubMed: 27743991]
- 39.
Su H, Lau YF. Identification of the transcriptional unit, structural organization, and promoter sequence of the human sex-determining region Y (SRY) gene, using a reverse genetic approach.
Am J Hum Genet. 1993 Jan;52(1):24-38. [
PMC free article: PMC1682107] [
PubMed: 8434602]
- 40.
Hossain A, Saunders GF. The human sex-determining gene SRY is a direct target of WT1.
J Biol Chem. 2001 May 18;276(20):16817-23. [
PubMed: 11278460]
- 41.
Massardier J, Roth P, Michel-Calemard L, Rudigoz RC, Bouvier R, Dijoud F, Arnould P, Combourieu D, Gaucherand P. Campomelic dysplasia: echographic suspicion in the first trimester of pregnancy and final diagnosis of two cases.
Fetal Diagn Ther. 2008;24(4):452-7. [
PubMed: 19033726]
- 42.
Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, Pasantes J, Bricarelli FD, Keutel J, Hustert E, Wolf U, Tommerup N, Schempp W, Scherer G. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9.
Cell. 1994 Dec 16;79(6):1111-20. [
PubMed: 8001137]
- 43.
Kim Y, Kobayashi A, Sekido R, DiNapoli L, Brennan J, Chaboissier MC, Poulat F, Behringer RR, Lovell-Badge R, Capel B. Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination.
PLoS Biol. 2006 Jun;4(6):e187. [
PMC free article: PMC1463023] [
PubMed: 16700629]
- 44.
De Santa Barbara P, Bonneaud N, Boizet B, Desclozeaux M, Moniot B, Sudbeck P, Scherer G, Poulat F, Berta P. Direct interaction of SRY-related protein SOX9 and steroidogenic factor 1 regulates transcription of the human anti-Müllerian hormone gene.
Mol Cell Biol. 1998 Nov;18(11):6653-65. [
PMC free article: PMC109250] [
PubMed: 9774680]
- 45.
Oba K, Yanase T, Nomura M, Morohashi K, Takayanagi R, Nawata H. Structural characterization of human Ad4bp (SF-1) gene.
Biochem Biophys Res Commun. 1996 Sep 04;226(1):261-7. [
PubMed: 8806624]
- 46.
Shen WH, Moore CC, Ikeda Y, Parker KL, Ingraham HA. Nuclear receptor steroidogenic factor 1 regulates the müllerian inhibiting substance gene: a link to the sex determination cascade.
Cell. 1994 Jun 03;77(5):651-61. [
PubMed: 8205615]
- 47.
Cate RL, Mattaliano RJ, Hession C, Tizard R, Farber NM, Cheung A, Ninfa EG, Frey AZ, Gash DJ, Chow EP. Isolation of the bovine and human genes for Müllerian inhibiting substance and expression of the human gene in animal cells.
Cell. 1986 Jun 06;45(5):685-98. [
PubMed: 3754790]
- 48.
Cohen-Haguenauer O, Picard JY, Mattéi MG, Serero S, Nguyen VC, de Tand MF, Guerrier D, Hors-Cayla MC, Josso N, Frézal J. Mapping of the gene for anti-müllerian hormone to the short arm of human chromosome 19.
Cytogenet Cell Genet. 1987;44(1):2-6. [
PubMed: 3028714]
- 49.
Behringer RR. The in vivo roles of müllerian-inhibiting substance.
Curr Top Dev Biol. 1994;29:171-87. [
PubMed: 7828438]
- 50.
Wang PY, Koishi K, McGeachie AB, Kimber M, Maclaughlin DT, Donahoe PK, McLennan IS. Mullerian inhibiting substance acts as a motor neuron survival factor in vitro.
Proc Natl Acad Sci U S A. 2005 Nov 08;102(45):16421-5. [
PMC free article: PMC1283469] [
PubMed: 16260730]
- 51.
Wang PY, Protheroe A, Clarkson AN, Imhoff F, Koishi K, McLennan IS. Müllerian inhibiting substance contributes to sex-linked biases in the brain and behavior.
Proc Natl Acad Sci U S A. 2009 Apr 28;106(17):7203-8. [
PMC free article: PMC2678437] [
PubMed: 19359476]
- 52.
Burkhardt E, Adham IM, Brosig B, Gastmann A, Mattei MG, Engel W. Structural organization of the porcine and human genes coding for a Leydig cell-specific insulin-like peptide (LEY I-L) and chromosomal localization of the human gene (INSL3).
Genomics. 1994 Mar 01;20(1):13-9. [
PubMed: 8020942]
- 53.
Adham IM, Burkhardt E, Benahmed M, Engel W. Cloning of a cDNA for a novel insulin-like peptide of the testicular Leydig cells.
J Biol Chem. 1993 Dec 15;268(35):26668-72. [
PubMed: 8253799]
- 54.
Klonisch T, Fowler PA, Hombach-Klonisch S. Molecular and genetic regulation of testis descent and external genitalia development.
Dev Biol. 2004 Jun 01;270(1):1-18. [
PubMed: 15136137]
- 55.
Shalet SM. Normal testicular function and spermatogenesis.
Pediatr Blood Cancer. 2009 Aug;53(2):285-8. [
PubMed: 19343782]
- 56.
Ulloa-Aguirre A, Reiter E, Crépieux P. FSH Receptor Signaling: Complexity of Interactions and Signal Diversity.
Endocrinology. 2018 Aug 01;159(8):3020-3035. [
PubMed: 29982321]
- 57.
Urbański HF, Kohama SG, Garyfallou VT. Mechanisms mediating the response of GnRH neurones to excitatory amino acids.
Rev Reprod. 1996 Sep;1(3):173-81. [
PubMed: 9414455]
- 58.
Kauffman AS, Clifton DK, Steiner RA. Emerging ideas about kisspeptin- GPR54 signaling in the neuroendocrine regulation of reproduction.
Trends Neurosci. 2007 Oct;30(10):504-11. [
PubMed: 17904653]
- 59.
Wildt L, Häusler A, Marshall G, Hutchison JS, Plant TM, Belchetz PE, Knobil E. Frequency and amplitude of gonadotropin-releasing hormone stimulation and gonadotropin secretion in the rhesus monkey.
Endocrinology. 1981 Aug;109(2):376-85. [
PubMed: 6788538]
- 60.
Savoy-Moore RT, Swartz KH. Several GnRH stimulation frequencies differentially release FSH and LH from isolated, perfused rat anterior pituitary cells.
Adv Exp Med Biol. 1987;219:641-5. [
PubMed: 3124522]
- 61.
Dufau ML. The luteinizing hormone receptor.
Annu Rev Physiol. 1998;60:461-96. [
PubMed: 9558473]
- 62.
Müller J, Skakkebaek NE. The prenatal and postnatal development of the testis.
Baillieres Clin Endocrinol Metab. 1992 Apr;6(2):251-71. [
PubMed: 1616445]
- 63.
Prince FP. Ultrastructural evidence of mature Leydig cells and Leydig cell regression in the neonatal human testis.
Anat Rec. 1990 Dec;228(4):405-17. [
PubMed: 2178325]
- 64.
Zirkin BR, Papadopoulos V. Leydig cells: formation, function, and regulation.
Biol Reprod. 2018 Jul 01;99(1):101-111. [
PMC free article: PMC6044347] [
PubMed: 29566165]
- 65.
Clermont Y. Two classes of spermatogonial stem cells in the monkey (Cercopithecus aethiops).
Am J Anat. 1969 Sep;126(1):57-71. [
PubMed: 4982042]
- 66.
Clermont Y, Antar M. Duration of the cycle of the seminiferous epithelium and the spermatogonial renewal in the monkey Macaca arctoides.
Am J Anat. 1973 Feb;136(2):153-65. [
PubMed: 4684877]
- 67.
Schulze C. Morphological characteristics of the spermatogonial stem cells in man.
Cell Tissue Res. 1979 May 18;198(2):191-9. [
PubMed: 466665]
- 68.
Cortes D, Müller J, Skakkebaek NE. Proliferation of Sertoli cells during development of the human testis assessed by stereological methods.
Int J Androl. 1987 Aug;10(4):589-96. [
PubMed: 3654012]
- 69.
Setchell BP. The Parkes Lecture. Heat and the testis.
J Reprod Fertil. 1998 Nov;114(2):179-94. [
PubMed: 10070346]
- 70.
EHRENBERG L, VON EHRENSTEIN G, HEDGRAN A. Gonad temperature and spontaneous mutation-rate in man.
Nature. 1957 Dec 21;180(4599):1433-4. [
PubMed: 13493561]
- 71.
Speit G. Effects of temperature on sister chromatid exchanges.
Hum Genet. 1980;55(3):333-6. [
PubMed: 7203467]
- 72.
Ritzén EM. Undescended testes: a consensus on management.
Eur J Endocrinol. 2008 Dec;159 Suppl 1:S87-90. [
PubMed: 18728121]
- 73.
Kanemoto K, Hayashi Y, Kojima Y, Maruyama T, Ito M, Kohri K. Accuracy of ultrasonography and magnetic resonance imaging in the diagnosis of non-palpable testis.
Int J Urol. 2005 Jul;12(7):668-72. [
PubMed: 16045560]
- 74.
Sarihan H, Sari A, Abeş M, Dinç H. Nonpalpable undescending testis. Value of magnetic resonance imaging.
Minerva Urol Nefrol. 1998 Dec;50(4):233-6. [
PubMed: 9973808]
- 75.
Maghnie M, Vanzulli A, Paesano P, Bragheri R, Palladini G, Preti P, Del Maschio A, Severi F. The accuracy of magnetic resonance imaging and ultrasonography compared with surgical findings in the localization of the undescended testis.
Arch Pediatr Adolesc Med. 1994 Jul;148(7):699-703. [
PubMed: 7912611]
- 76.
Schreck RR, Distèche C. Karyotyping.
Curr Protoc Hum Genet. 2001 May;Appendix 4:Appendix 4A. [
PubMed: 18428228]
- 77.
Cox MJ, Coplen DE, Austin PF. The incidence of disorders of sexual differentiation and chromosomal abnormalities of cryptorchidism and hypospadias stratified by meatal location.
J Urol. 2008 Dec;180(6):2649-52; discussion 2652. [
PubMed: 18951572]
- 78.
Skakkebaek NE, Rajpert-De Meyts E, Main KM. Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects.
Hum Reprod. 2001 May;16(5):972-8. [
PubMed: 11331648]
- 79.
Xing JS, Bai ZM. Is testicular dysgenesis syndrome a genetic, endocrine, or environmental disease, or an unexplained reproductive disorder?
Life Sci. 2018 Feb 01;194:120-129. [
PubMed: 29183799]
- 80.
van den Driesche S, Kolovos P, Platts S, Drake AJ, Sharpe RM. Inter-relationship between testicular dysgenesis and Leydig cell function in the masculinization programming window in the rat.
PLoS One. 2012;7(1):e30111. [
PMC free article: PMC3256232] [
PubMed: 22253897]
- 81.
Chen GR, Dong L, Ge RS, Hardy MP. [Relationship between phthalates and testicular dysgenesis syndrome].
Zhonghua Nan Ke Xue. 2007 Mar;13(3):195-200. [
PubMed: 17393778]
- 82.
He M, Yang C, Geng R, Zhao X, Hong L, Piao X, Chen T, Quinto M, Li D. Monitoring of phthalates in foodstuffs using gas purge microsyringe extraction coupled with GC-MS.
Anal Chim Acta. 2015 Jun 16;879:63-8. [
PubMed: 26002478]
- 83.
Cevasco A, Urbatzka R, Bottero S, Massari A, Pedemonte F, Kloas W, Mandich A. Endocrine disrupting chemicals (EDC) with (anti)estrogenic and (anti)androgenic modes of action affecting reproductive biology of Xenopus laevis: II. Effects on gonad histomorphology.
Comp Biochem Physiol C Toxicol Pharmacol. 2008 Mar;147(2):241-51. [
PubMed: 18032117]
- 84.
Mills LJ, Gutjahr-Gobell RE, Zaroogian GE, Horowitz DB, Laws SC. Modulation of aromatase activity as a mode of action for endocrine disrupting chemicals in a marine fish.
Aquat Toxicol. 2014 Feb;147:140-50. [
PubMed: 24418745]
- 85.
Troisi R, Hyer M, Hatch EE, Titus-Ernstoff L, Palmer JR, Strohsnitter WC, Herbst AL, Adam E, Hoover RN. Medical conditions among adult offspring prenatally exposed to diethylstilbestrol.
Epidemiology. 2013 May;24(3):430-8. [
PubMed: 23474687]
- 86.
Cabaton NJ, Canlet C, Wadia PR, Tremblay-Franco M, Gautier R, Molina J, Sonnenschein C, Cravedi JP, Rubin BS, Soto AM, Zalko D. Effects of low doses of bisphenol A on the metabolome of perinatally exposed CD-1 mice.
Environ Health Perspect. 2013 May;121(5):586-93. [
PMC free article: PMC3673190] [
PubMed: 23425943]
- 87.
Virtanen HE, Bjerknes R, Cortes D, Jørgensen N, Rajpert-De Meyts E, Thorsson AV, Thorup J, Main KM. Cryptorchidism: classification, prevalence and long-term consequences.
Acta Paediatr. 2007 May;96(5):611-6. [
PubMed: 17462053]
- 88.
Berkowitz GS, Lapinski RH, Dolgin SE, Gazella JG, Bodian CA, Holzman IR. Prevalence and natural history of cryptorchidism.
Pediatrics. 1993 Jul;92(1):44-9. [
PubMed: 8100060]
- 89.
Niedzielski JK, Oszukowska E, Słowikowska-Hilczer J. Undescended testis - current trends and guidelines: a review of the literature.
Arch Med Sci. 2016 Jun 01;12(3):667-77. [
PMC free article: PMC4889701] [
PubMed: 27279862]
- 90.
Suzuki Y, Sasagawa I, Itoh K, Ashida J, Ogata T. 5Alpha-reductase type 2 genes in Japanese males do not appear to be associated with cryptorchidism.
Fertil Steril. 2002 Aug;78(2):330-4. [
PubMed: 12137870]
- 91.
Silva-Ramos M, Oliveira JM, Cabeda JM, Reis A, Soares J, Pimenta A. The CAG repeat within the androgen receptor gene and its relationship to cryptorchidism.
Int Braz J Urol. 2006 May-Jun;32(3):330-4; discussion 335. [
PubMed: 16813680]
- 92.
Zimmermann S, Steding G, Emmen JM, Brinkmann AO, Nayernia K, Holstein AF, Engel W, Adham IM. Targeted disruption of the Insl3 gene causes bilateral cryptorchidism.
Mol Endocrinol. 1999 May;13(5):681-91. [
PubMed: 10319319]
- 93.
Nef S, Parada LF. Hormones in male sexual development.
Genes Dev. 2000 Dec 15;14(24):3075-86. [
PubMed: 11124800]
- 94.
Hutson JM, Hasthorpe S. Testicular descent and cryptorchidism: the state of the art in 2004.
J Pediatr Surg. 2005 Feb;40(2):297-302. [
PubMed: 15750918]
- 95.
Hutson JM, Li R, Southwell BR, Petersen BL, Thorup J, Cortes D. Germ cell development in the postnatal testis: the key to prevent malignancy in cryptorchidism?
Front Endocrinol (Lausanne). 2012;3:176. [
PMC free article: PMC3539691] [
PubMed: 23316184]
- 96.
Ivell R, Hartung S. The molecular basis of cryptorchidism.
Mol Hum Reprod. 2003 Apr;9(4):175-81. [
PubMed: 12651898]
- 97.
Wood HM, Elder JS. Cryptorchidism and testicular cancer: separating fact from fiction.
J Urol. 2009 Feb;181(2):452-61. [
PubMed: 19084853]
- 98.
Brauner R, Neve M, Allali S, Trivin C, Lottmann H, Bashamboo A, McElreavey K. Clinical, biological and genetic analysis of anorchia in 26 boys.
PLoS One. 2011;6(8):e23292. [
PMC free article: PMC3154292] [
PubMed: 21853106]
- 99.
Abeyaratne MR, Aherne WA, Scott JE. The vanishing testis.
Lancet. 1969 Oct 18;2(7625):822-4. [
PubMed: 4186283]
- 100.
Sarto GE, Opitz JM. The XY gonadal agenesis syndrome.
J Med Genet. 1973 Sep;10(3):288-93. [
PMC free article: PMC1013035] [
PubMed: 4774539]
- 101.
Vinci G, Anjot MN, Trivin C, Lottmann H, Brauner R, McElreavey K. An analysis of the genetic factors involved in testicular descent in a cohort of 14 male patients with anorchia.
J Clin Endocrinol Metab. 2004 Dec;89(12):6282-5. [
PubMed: 15579790]
- 102.
Philibert P, Zenaty D, Lin L, Soskin S, Audran F, Léger J, Achermann JC, Sultan C. Mutational analysis of steroidogenic factor 1 (NR5a1) in 24 boys with bilateral anorchia: a French collaborative study.
Hum Reprod. 2007 Dec;22(12):3255-61. [
PMC free article: PMC2990861] [
PubMed: 17940071]
- 103.
Fouatih K, Belin F, Lambert AS, Bouligand J, Bouvattier C. Pubertal growth spurt in patients with bilateral anorchia after testosterone replacement therapy.
Arch Pediatr. 2019 Sep;26(6):320-323. [
PubMed: 31353150]
- 104.
Smith NM, Byard RW, Bourne AJ. Testicular regression syndrome--a pathological study of 77 cases.
Histopathology. 1991 Sep;19(3):269-72. [
PubMed: 1916702]
- 105.
Szmer J, Chrzan R. Decision-making challenges in children with congenital and acquired monorchism: a critical literature review.
Folia Med Cracov. 2019;59(1):127-136. [
PubMed: 31180081]
- 106.
Pirgon Ö, Dündar BN. Vanishing testes: a literature review.
J Clin Res Pediatr Endocrinol. 2012 Sep;4(3):116-20. [
PMC free article: PMC3459158] [
PubMed: 22985611]
- 107.
Rozanski TA, Wojno KJ, Bloom DA. The remnant orchiectomy.
J Urol. 1996 Feb;155(2):712-3; discussion 714. [
PubMed: 8558712]
- 108.
Velázquez de Cuéllar Paracchi M, Leal Orozco A, Ruíz Serrano C, Aguado Roncero P, Pérez Tejerizo G, Soriano Guillén L. [Micropenis and bilateral cryptorchidism secondary to vanishing testes syndrome].
An Pediatr (Barc). 2009 Feb;70(2):199-200. [
PubMed: 19217585]
- 109.
Grinspon RP, Habib C, Bedecarrás P, Gottlieb S, Rey RA. Compensatory function of the remaining testis is dissociated in boys and adolescents with monorchidism.
Eur J Endocrinol. 2016 Mar;174(3):399-407. [
PubMed: 26671976]
- 110.
Uğuz S, Gürağaç A, Demirer Z, Yilmaz S, Aydur E. Bilateral polyorchidism with ipsilateral two undescended testes: a rare congenital anomaly.
Andrologia. 2017 May;49(4) [
PubMed: 27373456]
- 111.
Yeniyol CO, Nergiz N, Tuna A. Abdominal polyorchidism: a case report and review of the literature.
Int Urol Nephrol. 2004;36(3):407-8. [
PubMed: 15783116]
- 112.
Balasar M, Sönmez MG, Oltulu P, Kandemir A, Kılıç M, Göğer YE, Pişkin MM. Polyorchidism; unilateral, one atrophic undescended double testicles.
Urol Ann. 2017 Apr-Jun;9(2):208-210. [
PMC free article: PMC5405673] [
PubMed: 28479781]
- 113.
Jakhere SG, Saifi SA, Ranwaka AA. Supernumerary testis: Imaging appearance of a rare entity.
Indian J Urol. 2014 Apr;30(2):233-4. [
PMC free article: PMC3989832] [
PubMed: 24744529]
- 114.
Leung AK. Polyorchidism.
Am Fam Physician. 1988 Sep;38(3):153-6. [
PubMed: 3046267]
- 115.
Favorito LA, Cavalcante AG, Costa WS. Anatomic aspects of epididymis and tunica vaginalis in patients with testicular torsion.
Int Braz J Urol. 2004 Sep-Oct;30(5):420-4. [
PubMed: 15610580]
- 116.
Castilla EE, Sod R, Anzorena O, Texido J. Neonatal testicular torsion in two brothers.
J Med Genet. 1975 Mar;12(1):112-3. [
PMC free article: PMC1013243] [
PubMed: 1121017]
- 117.
Collins K, Broecker BH. Familial torsion of the spermatic cord.
J Urol. 1989 Jan;141(1):128-9. [
PubMed: 2908936]
- 118.
CUNNINGHAM RF. Familial occurrence of testicular torsion.
JAMA. 1960 Nov 05;174:1330-1. [
PubMed: 13718864]
- 119.
Martin AD, Rushton HG. The prevalence of bell clapper anomaly in the solitary testis in cases of prior perinatal torsion.
J Urol. 2014 May;191(5 Suppl):1573-7. [
PubMed: 24679875]
- 120.
Monteilh C, Calixte R, Burjonrappa S. Controversies in the management of neonatal testicular torsion: A meta-analysis.
J Pediatr Surg. 2019 Apr;54(4):815-819. [
PubMed: 30098810]
- 121.
Riaz-Ul-Haq M, Mahdi DE, Elhassan EU. Neonatal testicular torsion; a review article.
Iran J Pediatr. 2012 Sep;22(3):281-9. [
PMC free article: PMC3564080] [
PubMed: 23400637]
- 122.
Da Silva Rios S, Monteiro IC, Braz Dos Santos LG, Caldas NG, Chen AC, Chen JR, Silva HS. A case of swyer syndrome associated with advanced gonadal dysgerminoma involving long survival.
Case Rep Oncol. 2015 Jan-Apr;8(1):179-84. [
PMC free article: PMC4410511] [
PubMed: 25960730]
- 123.
Lipay MV, Bianco B, Verreschi IT. [Gonadal dysgenesis and tumors: genetic and clinical features].
Arq Bras Endocrinol Metabol. 2005 Feb;49(1):60-70. [
PubMed: 16544035]
- 124.
King TF, Conway GS. Swyer syndrome.
Curr Opin Endocrinol Diabetes Obes. 2014 Dec;21(6):504-10. [
PubMed: 25314337]
- 125.
Nieschlag E. Klinefelter syndrome: the commonest form of hypogonadism, but often overlooked or untreated.
Dtsch Arztebl Int. 2013 May;110(20):347-53. [
PMC free article: PMC3674537] [
PubMed: 23825486]
- 126.
Kumar P, Kumar N, Thakur DS, Patidar A. Male hypogonadism: Symptoms and treatment.
J Adv Pharm Technol Res. 2010 Jul;1(3):297-301. [
PMC free article: PMC3255409] [
PubMed: 22247861]
- 127.
Tüttelmann F, Gromoll J. Novel genetic aspects of Klinefelter's syndrome.
Mol Hum Reprod. 2010 Jun;16(6):386-95. [
PubMed: 20228051]
- 128.
Maiburg M, Repping S, Giltay J. The genetic origin of Klinefelter syndrome and its effect on spermatogenesis.
Fertil Steril. 2012 Aug;98(2):253-60. [
PubMed: 22749222]
- 129.
Batista RL, Costa EMF, Rodrigues AS, Gomes NL, Faria JA, Nishi MY, Arnhold IJP, Domenice S, Mendonca BB. Androgen insensitivity syndrome: a review.
Arch Endocrinol Metab. 2018 Mar-Apr;62(2):227-235. [
PMC free article: PMC10118986] [
PubMed: 29768628]
- 130.
Mendonca BB, Costa EM, Belgorosky A, Rivarola MA, Domenice S. 46,XY DSD due to impaired androgen production.
Best Pract Res Clin Endocrinol Metab. 2010 Apr;24(2):243-62. [
PubMed: 20541150]
- 131.
Melo KF, Mendonça BB, Billerbeck AE, Costa EM, Latronico AC, Arnhold IJ. [Androgen insensitivity syndrome: clinical, hormonal and molecular analysis of 33 cases].
Arq Bras Endocrinol Metabol. 2005 Feb;49(1):87-97. [
PubMed: 16544039]
- 132.
Hughes IA, Deeb A. Androgen resistance.
Best Pract Res Clin Endocrinol Metab. 2006 Dec;20(4):577-98. [
PubMed: 17161333]
- 133.
Tan MH, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: structure, role in prostate cancer and drug discovery.
Acta Pharmacol Sin. 2015 Jan;36(1):3-23. [
PMC free article: PMC4571323] [
PubMed: 24909511]
- 134.
Mongan NP, Tadokoro-Cuccaro R, Bunch T, Hughes IA. Androgen insensitivity syndrome.
Best Pract Res Clin Endocrinol Metab. 2015 Aug;29(4):569-80. [
PubMed: 26303084]
- 135.
Gulía C, Baldassarra S, Zangari A, Briganti V, Gigli S, Gaffi M, Signore F, Vallone C, Nucciotti R, Costantini FM, Pizzuti A, Bernardo S, Porrello A, Piergentili R. Androgen insensitivity syndrome.
Eur Rev Med Pharmacol Sci. 2018 Jun;22(12):3873-3887. [
PubMed: 29949163]
- 136.
Cheon CK. Practical approach to steroid 5alpha-reductase type 2 deficiency.
Eur J Pediatr. 2011 Jan;170(1):1-8. [
PubMed: 20349245]
- 137.
Mendonca BB, Domenice S, Arnhold IJ, Costa EM. 46,XY disorders of sex development (DSD).
Clin Endocrinol (Oxf). 2009 Feb;70(2):173-87. [
PubMed: 18811725]
- 138.
Speiser PW. Congenital Adrenal Hyperplasia.
F1000Res. 2015;4(F1000 Faculty Rev):601. [
PMC free article: PMC4544382] [
PubMed: 26339484]
- 139.
Speiser PW, White PC. Congenital adrenal hyperplasia.
N Engl J Med. 2003 Aug 21;349(8):776-88. [
PubMed: 12930931]
- 140.
Witchel SF. Congenital Adrenal Hyperplasia.
J Pediatr Adolesc Gynecol. 2017 Oct;30(5):520-534. [
PMC free article: PMC5624825] [
PubMed: 28450075]
- 141.
Simard J, Moisan AM, Morel Y. Congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase/Delta(5)-Delta(4) isomerase deficiency.
Semin Reprod Med. 2002 Aug;20(3):255-76. [
PubMed: 12428206]
- 142.
Krone N, Dhir V, Ivison HE, Arlt W. Congenital adrenal hyperplasia and P450 oxidoreductase deficiency.
Clin Endocrinol (Oxf). 2007 Feb;66(2):162-72. [
PubMed: 17223983]
- 143.
New MI. Male pseudohermaphroditism due to 17 alpha-hydroxylase deficiency.
J Clin Invest. 1970 Oct;49(10):1930-41. [
PMC free article: PMC322683] [
PubMed: 5456802]
- 144.
Hauffa BP, Miller WL, Grumbach MM, Conte FA, Kaplan SL. Congenital adrenal hyperplasia due to deficient cholesterol side-chain cleavage activity (20, 22-desmolase) in a patient treated for 18 years.
Clin Endocrinol (Oxf). 1985 Nov;23(5):481-93. [
PubMed: 3841304]
- 145.
Bose HS, Sugawara T, Strauss JF, Miller WL., International Congenital Lipoid Adrenal Hyperplasia Consortium. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia.
N Engl J Med. 1996 Dec 19;335(25):1870-8. [
PubMed: 8948562]
- 146.
Metherell LA, Naville D, Halaby G, Begeot M, Huebner A, Nürnberg G, Nürnberg P, Green J, Tomlinson JW, Krone NP, Lin L, Racine M, Berney DM, Achermann JC, Arlt W, Clark AJ. Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency.
J Clin Endocrinol Metab. 2009 Oct;94(10):3865-71. [
PMC free article: PMC2860769] [
PubMed: 19773404]
- 147.
Primatesta P, Goldacre MJ. Inguinal hernia repair: incidence of elective and emergency surgery, readmission and mortality.
Int J Epidemiol. 1996 Aug;25(4):835-9. [
PubMed: 8921464]
- 148.
Ruhl CE, Everhart JE. Risk factors for inguinal hernia among adults in the US population.
Am J Epidemiol. 2007 May 15;165(10):1154-61. [
PubMed: 17374852]
- 149.
Tanyel FC, Müftüoglu S, Dagdeviren A, Kaymaz FF, Büyükpamukçu N. Myofibroblasts defined by electron microscopy suggest the dedifferentiation of smooth muscle within the sac walls associated with congenital inguinal hernia.
BJU Int. 2001 Feb;87(3):251-5. [
PubMed: 11167652]
- 150.
Rodprasert W, Virtanen HE, Mäkelä JA, Toppari J. Hypogonadism and Cryptorchidism.
Front Endocrinol (Lausanne). 2019;10:906. [
PMC free article: PMC6974459] [
PubMed: 32010061]
- 151.
Foresta C, Zuccarello D, Garolla A, Ferlin A. Role of hormones, genes, and environment in human cryptorchidism.
Endocr Rev. 2008 Aug;29(5):560-80. [
PubMed: 18436703]
- 152.
Ramareddy RS, Alladi A, Siddappa OS. Ectopic testis in children: experience with seven cases.
J Pediatr Surg. 2013 Mar;48(3):538-41. [
PubMed: 23480908]
- 153.
Ferro F, Lais A, Matarazzo E, Capozza N, Caione P. [Retractile testis and gliding testis. Two distinct clinical entities].
Minerva Urol Nefrol. 1996 Sep;48(3):145-9. [
PubMed: 8966651]
- 154.
Cryptorchidism: a prospective study of 7500 consecutive male births, 1984-8. John Radcliffe Hospital Cryptorchidism Study Group.
Arch Dis Child. 1992 Jul;67(7):892-9. [
PMC free article: PMC1793845] [
PubMed: 1355643]
- 155.
Barteczko KJ, Jacob MI. The testicular descent in human. Origin, development and fate of the gubernaculum Hunteri, processus vaginalis peritonei, and gonadal ligaments.
Adv Anat Embryol Cell Biol. 2000;156:III-X, 1-98. [
PubMed: 11008363]
- 156.
Nguyen HT, Coakley F, Hricak H. Cryptorchidism: strategies in detection.
Eur Radiol. 1999;9(2):336-43. [
PubMed: 10101659]
- 157.
Borrow M, Gough MH. Bilateral absence of testes.
Lancet. 1970 Feb 14;1(7642):366. [
PubMed: 4189631]
- 158.
Aynsley-Green A, Zachmann M, Illig R, Rampini S, Prader A. Congenital bilateral anorchia in childhood: a clinical, endocrine and therapeutic evaluation of twenty-one cases.
Clin Endocrinol (Oxf). 1976 Jul;5(4):381-91. [
PubMed: 786507]
- 159.
Zenaty D, Dijoud F, Morel Y, Cabrol S, Mouriquand P, Nicolino M, Bouvatier C, Pinto G, Lecointre C, Pienkowski C, Soskin S, Bost M, Bertrand AM, El-Ghoneimi A, Nihoul-Fekete C, Léger J. Bilateral anorchia in infancy: occurence of micropenis and the effect of testosterone treatment.
J Pediatr. 2006 Nov;149(5):687-91. [
PubMed: 17095345]
- 160.
De Marchi M, Campagnoli C, Ghiringhello B, Ponzio G, Carbonara A. Gonadal agenesis in a phenotypically normal female with positive H-Y antigen.
Hum Genet. 1981;56(3):417-9. [
PubMed: 7239525]
- 161.
Shadpour P, Kashi AH, Arvin A. Scrotal testis size in unilateral non-palpable cryptorchidism, what it can and cannot tell: Study of a Middle Eastern population.
J Pediatr Urol. 2017 Jun;13(3):268.e1-268.e6. [
PubMed: 28254240]
- 162.
Hodhod A, Capolicchio JP, Jednak R, El-Sherbiny M. Testicular hypertrophy as a predictor for contralateral monorchism: Retrospective review of prospectively recorded data.
J Pediatr Urol. 2016 Feb;12(1):34.e1-5. [
PubMed: 26279100]
- 163.
Kheirandish P, Chinegwundoh F. An unusual case of triorchidism.
JRSM Short Rep. 2010 Nov 25;1(6):55. [
PMC free article: PMC2994354] [
PubMed: 21234118]
- 164.
Arlen AM, Holzman SA, Weiss AD, Garola RE, Cerwinka WH. Functional supernumerary testis in a child with testicular torsion and review of polyorchidism.
Pediatr Surg Int. 2014 May;30(5):565-8. [
PubMed: 24557155]
- 165.
Abduljabbar AH. A Case Report: Triorchidism; is a Rare Mistaken Cause for Extra Testicular Neoplasm.
Urol Case Rep. 2015 May;3(3):89-91. [
PMC free article: PMC4714288] [
PubMed: 26793513]
- 166.
Kharrazi SM, Rahmani MR, Sakipour M, Khoob S. Polyorchidism: a case report and review of literature.
Urol J. 2006 Summer;3(3):180-3. [
PubMed: 17559037]
- 167.
Bergholz R, Wenke K. Polyorchidism: a meta-analysis.
J Urol. 2009 Nov;182(5):2422-7. [
PubMed: 19765760]
- 168.
Zhao LC, Lautz TB, Meeks JJ, Maizels M. Pediatric testicular torsion epidemiology using a national database: incidence, risk of orchiectomy and possible measures toward improving the quality of care.
J Urol. 2011 Nov;186(5):2009-13. [
PubMed: 21944120]
- 169.
Huang WY, Chen YF, Chang HC, Yang TK, Hsieh JT, Huang KH. The incidence rate and characteristics in patients with testicular torsion: a nationwide, population-based study.
Acta Paediatr. 2013 Aug;102(8):e363-7. [
PubMed: 23611668]
- 170.
Lee SM, Huh JS, Baek M, Yoo KH, Min GE, Lee HL, Lee DG. A nationwide epidemiological study of testicular torsion in Korea.
J Korean Med Sci. 2014 Dec;29(12):1684-7. [
PMC free article: PMC4248591] [
PubMed: 25469070]
- 171.
Hyun GS. Testicular Torsion.
Rev Urol. 2018;20(2):104-106. [
PMC free article: PMC6168322] [
PubMed: 30288149]
- 172.
Yiee JH, Chang L, Kaplan A, Kwan L, Chung PJ, Litwin MS. Patterns of care in testicular torsion: influence of hospital transfer on testicular outcomes.
J Pediatr Urol. 2013 Dec;9(6 Pt A):713-20. [
PMC free article: PMC3999916] [
PubMed: 23896260]
- 173.
Pogorelić Z, Mrklić I, Jurić I, Biočić M, Furlan D. Testicular torsion in the inguinal canal in children.
J Pediatr Urol. 2013 Dec;9(6 Pt A):793-7. [
PubMed: 23123082]
- 174.
Yazbeck S, Patriquin HB. Accuracy of Doppler sonography in the evaluation of acute conditions of the scrotum in children.
J Pediatr Surg. 1994 Sep;29(9):1270-2. [
PubMed: 7807366]
- 175.
Wikström AM, Hoei-Hansen CE, Dunkel L, Rajpert-De Meyts E. Immunoexpression of androgen receptor and nine markers of maturation in the testes of adolescent boys with Klinefelter syndrome: evidence for degeneration of germ cells at the onset of meiosis.
J Clin Endocrinol Metab. 2007 Feb;92(2):714-9. [
PubMed: 17148558]
- 176.
Avendaño A, Paradisi I, Cammarata-Scalisi F, Callea M. 5-α-Reductase type 2 deficiency: is there a genotype-phenotype correlation? A review.
Hormones (Athens). 2018 Jun;17(2):197-204. [
PubMed: 29858846]
- 177.
Byne W. Developmental endocrine influences on gender identity: implications for management of disorders of sex development.
Mt Sinai J Med. 2006 Nov;73(7):950-9. [
PubMed: 17195880]
- 178.
Thilén A, Larsson A. Congenital adrenal hyperplasia in Sweden 1969-1986. Prevalence, symptoms and age at diagnosis.
Acta Paediatr Scand. 1990 Feb;79(2):168-75. [
PubMed: 2321478]
- 179.
Bois E, Mornet E, Chompret A, Feingold J, Hochez J, Goulet V. [Congenital adrenal hyperplasia (21-OH) in France. Population genetics].
Arch Fr Pediatr. 1985 Mar;42(3):175-9. [
PubMed: 3873926]
- 180.
Yanase T, Simpson ER, Waterman MR. 17 alpha-hydroxylase/17,20-lyase deficiency: from clinical investigation to molecular definition.
Endocr Rev. 1991 Feb;12(1):91-108. [
PubMed: 2026124]
Disclosure: Owuraku Titi-Lartey declares no relevant financial relationships with ineligible companies.
Disclosure: Yusuf Khan declares no relevant financial relationships with ineligible companies.