NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002-.

This publication is provided for historical reference only and the information may be out of date.
- 3-D or 3D
Three-dimensional.
- Accession number
An Accession number is a unique identifier given to a sequence when it is submitted to one of the DNA repositories (GenBank, EMBL, DDBJ). The initial deposition of a sequence record is referred to as version 1. If the sequence is updated, the version number is incremented, but the Accession number will remain constant.
- Alu
The Alu repeat family comprises short interspersed elements (SINES) present in multiple copies in the genomes of humans and other primates. The Alu sequence is approximately 300 bp in length and is found commonly in introns, 3′ untranslated regions of genes, and intergenic genomic regions. They are mobile elements and are present in the human genome in extremely high copy number. Almost 1 million copies of the Alu sequence are estimated to be present, making it the most abundant mobile element. The Alu sequence is so named because of the presence of a recognition site for the AluI endonuclease in the middle of the Alu sequence. Because of the widespread occurrence of the Alu repeat in the genome, the Alu sequence is used as a universal primer for PCR in animal cell lines; it binds in both forward and reverse directions. The Alu universal primer sequence is as follows: 5′-GTG GAT CAC CTG AGG TCA GGA GTT TC-3′ (26-mer).
- allele
One of the variant forms of a gene at a particular locus on a chromosome. Different alleles produce variation in inherited characteristics such as hair color or blood type. In an individual, one form of the allele (the dominant one) may be expressed more than another form (the recessive one). When “genes” are considered simply as segments of a nucleotide sequence, allele refers to each of the possible alternative nucleotides at a specific position in the sequence. For example, a CT polymorphism such as CCT[C/T]CCAT would have two alleles: C and T.
- API
Application Programming Interface. An API is a set of routines that an application uses to request and carry out lower-level services performed by a computer's operating system. For computers running a graphical user interface, an API manages an application's windows, icons, menus, and dialog boxes.
- ASN.1
Abstract Syntax Notation 1 is an international standard data-representation format used to achieve interoperability between computer platforms. It allows for the reliable exchange of data in terms of structure and content by computer and software systems of all types.
- BAC
Bacterial Artificial Chromosome. A BAC is a large segment of DNA (100,000–200,000 bp) from another species cloned into bacteria. Once the foreign DNA has been cloned into the host bacteria, many copies of it can be made.
- BankIt
BankIt is a tool for the online submission of one or a few sequences into GenBank and is designed to make the submission process quick and easy. (BankIt also automatically uses VecScreen to identify segments of nucleic acid sequence that may be of vector, adapter, or linker origin to combat the problem of vector contamination in GenBank.)
- bit score
The value S′ is derived from the raw alignment score S in which the statistical properties of the scoring system used have been taken into account. By normalizing a raw score using the formula:
a “bit score” S′ is attained, which has a standard set of units, and where K and lambda are the statistical parameters of the scoring system. Because bit scores have been normalized with respect to the scoring system, they can be used to compare alignment scores from different searches.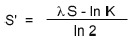
- BLAST
Basic Local Alignment Search Tool (Altschul et al., J Mol Biol 215:403-410; 1990). A sequence comparison algorithm that is optimized for speed and used to search sequence databases for optimal local alignments to a query. See the BLAST chapter (Chapter 15) or the tutorial or the narrative guide to BLAST.
- blastn
nucleotide–nucleotide BLAST. blastn takes nucleotide sequences in FASTA format, GenBank Accession numbers, or GI numbers and compares them against the NCBI Nucleotide databases.
- blastp
protein–protein BLAST. blastp takes protein sequences in FASTA format, GenBank Accession numbers, or GI numbers and compares them against the NCBI Protein databases.
- BLAT
A DNA/Protein sequence analysis program to quickly find sequences of 95% and greater similarity of length 40 bases or more. It may miss more divergent or shorter sequence alignments. BLAT on proteins finds sequences of 80% and greater similarity of length 20 amino acids or more. BLAT is not BLAST. (See the BLAT web page.)
- BLink
BLAST Link. BLink displays the results of BLAST searches that have been done for every protein sequence in the Entrez Protein data domain. It can be accessed by following the BLink link displayed beside any hit in the results of an Entrez Protein search. In contrast to Entrez's Related Sequences feature, which lists the titles of similar sequences, BLink displays the graphical output of precomputed blastp results against the non-redundant (nr) protein database. The output includes the positions of up to 200 BLAST hits on the query sequence, scores, and alignments. BLink offers a variety of display options, including the distribution of hits by taxonomic grouping, the best hit to each organism, the protein domains in the query sequence, similar sequences that have known 3D structures, and more. Additional options allow you to specify from which taxa you would like to exclude, increase, or decrease the BLAST cutoff score or filter the BLAST hits to show only those from a specific source database, such as RefSeq or SWISS-PROT. See the BLink help document for additional information.
- BLOB
Binary Large Object (or binary data object). BLOB refers to a large piece of data, such as a bitmap. A BLOB is characterized by large field values, an unpredictable table size, and data that are formless from the perspective of a program. It is also a keyword designating the BLOB structure, which contains information about a block of data.
- BLOSUM 62
Blocks Substitution Matrix. A substitution matrix in which scores for each position are derived from observations of the frequencies of substitutions in blocks of local alignments in related proteins. Each matrix is tailored to a particular evolutionary distance. In the BLOSUM 62 matrix, for example, the alignment from which scores were derived was created using sequences sharing no more than 62% identity. Sequences more identical than 62% are represented by a single sequence in the alignment to avoid overweighting closely related family members (Henikoff and Henikoff, Proc Natl Acad Sci U S A 89:10915-10919; 1992).
- Boolean
This term refers to binary algebra that uses the logical operators AND, OR, XOR, and NOT; the outcomes consist of logical values (either TRUE or FALSE). The keyword boolean indicates that the expression or constant expression associated with the identifier takes the value TRUE or FALSE. The logical-AND (&&) operator produces the value 1 if both operands have nonzero values; otherwise, it produces the value 0. The logical-OR (׀׀) operator produces the value 1 if either of its operands has a nonzero value. The logical-NOT (!) operator produces the value 0 if its operand is true (nonzero) and the value 1 if its operand is FALSE (0). The exclusive OR (XOR) operator yields TRUE only if one of its operands are TRUE and the other is FALSE. If both operands are the same (either TRUE or FALSE), the operation yields FALSE.
- build
A run of the genome assembly and annotation process of the set of products generated by that run.
- CCAP
Cancer Chromosome Aberration Project. CCAP was designed to expedite the definition and detailed characterization of the distinct chromosomal alterations that are associated with malignant transformation. The project is a collaboration among the NCI, the NCBI, and numerous research labs.
- CD
Conserved Domain. CD refers to a domain (a distinct functional and/or structural unit of a protein) that has been conserved during evolution. During evolution, changes at specific positions of an amino acid sequence in the protein have occurred in a way that preserve the physico-chemical properties of the original residues, and hence the structural and/or functional properties of that region of the protein.
- CDART
Conserved Domain Architecture Retrieval Tool. When given a protein query sequence, CDART displays the functional domains that make up the protein and lists proteins with similar domain architectures. The functional domains for a sequence are found by comparing the protein sequence to a database of conserved domain alignments, CDD using RPS-BLAST.
- CDD
Conserved Domain Database. This database is a collection of sequence alignments and profiles representing protein domains conserved during molecular evolution.
- cDNA
complementary DNA. A DNA sequence obtained by reverse transcription of a messenger RNA (mRNA) sequence.
- CDS
coding region, coding sequence. CDS refers to the portion of a genomic DNA sequence that is translated, from the start codon to the stop codon, inclusively, if complete. A partial CDS lacks part of the complete CDS (it may lack either or both the start and stop codons). Successful translation of a CDS results in the synthesis of a protein.
- CEPH
- CGAP
Cancer Genome Anatomy Project. CGAP is an interdisciplinary program to identify the human genes expressed in different cancerous states, based on cDNA (EST) libraries, and to determine the molecular profiles of normal, precancerous, and malignant cells. The project is a collaboration among the NCI, the NCBI, and numerous research labs.
- CGH
Comparative Genomic Hybidization. CGH is a fluorescent molecular cytogenetic technique that identifies chromosomal aberrations and maps these changes to metaphase chromosomes. CGH can be used to generate a map of DNA copy number changes in tumor genomes. CGH is based on quantitative two-color fluorescence in situ hybridization (FISH). DNA extracted from tumor cells is labeled in one color (e.g., green) and mixed in a 1:1 ratio with DNA from normal cells, which is labeled in a different color (e.g., red). The mixture is then applied to normal metaphase chromosomes. Portions of the genome that are equally represented in normal and tumor cells will appear orange, regions that are deleted in the tumor sample relative to the normal sample will appear red, and regions that are present in higher copy number in the tumor sample (because of amplification) will appear green. Special image analysis tools are necessary to quantitate the ratio of green-to-red fluorescence to determine whether a given region is more highly represented in the normal or in the tumor sample.
- CGI
Common Gateway Interface. A mechanism that allows a Web server to run a program or script on the server and send the output to a Web browser.
- cluster
A group that is created based on certain criteria. For example, a gene cluster may include a set of genes whose similar expression profiles are found to be similar according to certain criteria, or a cluster may refer to a group of clones that are related to each other by homology.
- Cn3D
“See in 3-D” is a structure and sequence alignment viewer for NCBI databases. It allows viewing of 3-D structures and sequence–structure or structure–structure alignments. Cn3D can work as a helper application to the browser or as a client–server application that retrieves structure records from the Molecular Modeling Database (MMDB, see below) directly from the internet. The Cn3D homepage provides access to information on how to install the program, a tutorial to get started, and a comprehensive help document.
- codon
Sequence of three nucleotides in DNA or mRNA that specifies a particular amino acid during protein synthesis; also called a triplet. Of the 64 possible codons, 3 are stop codons, which do not specify amino acids.
- COGs
Clusters of Orthologous Groups (of proteins) were delineated by comparing protein sequences from completely sequenced genomes. Each COG consists of individual proteins or groups of paralogs from at least three lineages and thus corresponds to an ancient conserved domain.
- consensus sequence
The nucleotides or amino acids found most commonly at each position in the sequences of homologous DNAs, RNAs, or proteins.
- contig
A contiguous segment of the genome made by joining overlapping clones or sequences. A clone contig consists of a group of cloned (copied) pieces of DNA representing overlapping regions of a particular chromosome. A sequence contig is an extended sequence created by merging primary sequences that overlap. A contig map shows the regions of a chromosome where contiguous DNA segments overlap. Contig maps provide the ability to study a complete and often large segment of the genome by examining a series of overlapping clones, which then provide an unbroken succession of information about that region.
- Coriell
- CPU
Central Processing Unit. The CPU is the computational and control unit of a computer, the device that interprets and executes instructions.
- CSS
Cascading Style Sheets. CSS specify the formatting details that control the presentation and layout of HTML and XML elements. CSS can be used for describing the formatting behavior and text decoration of simply structured XML documents but cannot display structure that varies from the structure of the source data.
- Cubby
A tool of Entrez, the Cubby stores search strategies that may be updated at any time, stores LinkOut preferences to specify which LinkOut providers have to be displayed in PubMed, and changes the default document delivery service.
- DCMS
Data Creation and Maintenance System
- DDBJ
- definition line
A sequence in FASTA format begins with a single-line description, followed by lines of sequence data. The definition line or description line is distinguished from the sequence data by a “greater than” (>) symbol in the first column (see example); also DEFLINE, as in a flatfile.
- DNA
Deoxyribonucleic acid is the chemical inside the nucleus of a cell that carries the genetic instructions for making living organisms. DNA is composed of two anti-parallel strands, each a linear polymer of nucleotides. Each nucleotide has a phosphate group linked by a phosphoester bond to a pentose (a five-carbon sugar molecule, deoxyribose), that in turn is linked to one of four organic bases, adenine, guanine, cytosine, or thymine, abbreviated A, G, C, and T, respectively. The bases are of two types: purines, which have two rings and are slightly larger (A and G); and pyrimidines, which have only one ring (C and T). Each nucleotide is joined to the next nucleotide in the chain by a covalent phosphodiester bond between the 5′ carbon of one deoxyribose group and the 3′ carbon of the next. DNA is a helical molecule with the sugar–phosphate backbone on the outside and the nucleotides extending toward the central axis. There is specific base-pairing between the bases on opposite strands in such a way that A always pairs with T and G always pairs with C.
- domain
A “domain” refers to a discrete portion of a protein assumed to fold independently of the rest of the protein and which possesses its own function.
- draft sequence
Draft sequence refers to DNA sequence that is not yet finished but is generally of high quality (i.e., an accuracy of greater than 90%). Draft sequence data are mostly in the form of 10,000 base pair-sized fragments, the approximate chromosomal locations of which are known. The following keywords are associated with draft sequence: phase 0, light-pass coverage of a clone, generally only 1× coverage; phase 1, 4–10× coverage of a BAC clone (order and orientation of the fragments are unknown); and phase 2, 4–10× coverage of a BAC clone (order and orientation of the fragments are known). Phase 3 refers to the completely finished sequence.
- DTD
Document Type Definition. The DTD is an optional part of the prolog of an XML document that defines the rules of the document. It sets constraints for an XML document by specifying which elements are present in the document and the relationships between elements, e.g., which tags can contain other tags, the number and sequence of the tags, and attributes of the tags. The DTD helps to validate the data when the receiving application does not have a built-in description of the incoming data.
- DUST
A program for filtering low-complexity regions from nucleic acid sequences.
- E-value
Expect value. The E-value is a parameter that describes the number of hits one can “expect” to see by chance when searching a database of a particular size. It decreases exponentially with the score (S) that is assigned to a match between two sequences. Essentially, the E-value describes the random background noise that exists for matches between sequences. For example, an E-value of 1 assigned to a hit can be interpreted as meaning that in a database of the current size, one might expect to see one match with a similar score simply by chance. This means that the lower the E-value, or the closer it is to “0”, the higher is the “significance” of the match. However, it is important to note that searches with short sequences can be virtually identical and have relatively high E-value. This is because the calculation of the E-value also takes into account the length of the query sequence. This is because shorter sequences have a high probability of occurring in the database purely by chance. For more information, see the following tutorial.
- EC number
A number assigned to a type of enzyme according to a scheme of standardized enzyme nomenclature developed by the Enzyme Commission of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (IUBMB). EC numbers may be found in ENZYME, the Enzyme nomenclature database, maintained at the ExPASy molecular biology server.
- EMBL
- Entrez
Entrez is a retrieval system for searching several linked databases. It provides access to the following NCBI databases: PubMed, GenBank, Protein, Structure, Genome, PopSet, OMIM, Taxonomy, Books, ProbeSet, 3D Domains, UniSTS, SNP, and CDD. (See the Entrez chapter or the Entrez web page.)
- Entrez Gene
(formerly known as LocusLink). Entrez Gene provides tracked, unique identifiers for genes (GeneIDs) and reports information associated with those identifiers for unrestricted public use. See the Entrez Gene chapter or web page.)
- EST
Expressed Sequence Tag. ESTs are short (usually approximately 300–500 base pairs), single-pass sequence reads from cDNA. Typically, they are produced in large batches. They represent the genes expressed in a given tissue and/or at a given developmental stage. They are tags (some coding, others not) of expression for a given cDNA library. They are useful in identifying full-length genes and in mapping.
- e-PCR
Electronic PCR is used to compare a query sequence to mapped sequence-tagged sites (STSs) to find a possible map location for the query sequence. e-PCR finds STSs in DNA sequences by searching for subsequences that closely match the PCR primers present in mapped markers. The subsequences must have the correct order, orientation, and spacing that they could plausibly prime the amplification of a PCR product of the correct molecular weight.
- epub citation
“Ahead-of-print” citation. PubMed now accepts citations from publishers for articles that have been published electronically ahead of the printed issue. PubMed displays the category “[epub ahead of print]” in the part of the citation where the volume and pagination would ordinarily display. For example: Proc Natl Acad Sci U S A. 2000 May 2 [epub ahead of print].
- ExoFish
Exon Finding by Sequence Homology. Exofish is a tool based on homology searches for the rapid and reliable identification of human genes. It relies on the sequence of another vertebrate, the pufferfish Tetraodon nigroviridis (similar to Fugu), to detect conserved sequences with a very low background. The genome of T. nigroviridis is eight times more compact than the human genome and has been used in the comparative identification of human genes from the rough draft of the human genome (Roest Crollius et al., Nat Genet 25:235-238; 2000).
- exon
Refers to the portion of a gene that encodes for a part of that gene's mRNA. A gene may comprise many exons, some of which may include only protein-coding sequence; however, an exon may also include 5' or 3' untranslated sequence. Each exon codes for a specific portion of the complete protein. In some species (including humans), a gene's exons are separated by long regions of DNA (called introns or sometimes “junk DNA”) that often have no apparent function but have been shown to encode small untranslated RNAs or regulatory information. (See also splice sites.)
- exon-trapped
Exon trapping is a technique for cloning exon sequences from genomic DNA by selecting for functional splice sites, relying on the cellular splicing machinery. The genomic DNA containing the putative exon(s) is cloned into an exon-trap vector, which has a promoter, polyadenylation signals, and splice sites, and then transfected into a cell line. If there are functional splice sites in the genomic DNA fragment, the segments of DNA between the splice sites will be removed. Total RNA is isolated and reverse-transcribed. After cDNA synthesis and PCR amplification, the exon of interest is cloned.
- ExPASy
Expert Protein Analysis System is a proteomics server of the Swiss Bioinformatics Institute (SIB).
- FASTA
The first widely used algorithm for similarity searching of protein and DNA sequence databases. The program looks for optimal local alignments by scanning the sequence for small matches called “words”. Initially, the scores of segments in which there are multiple word hits are calculated (“init1”). Later, the scores of several segments may be summed to generate an “initn” score. An optimized alignment that includes gaps is shown in the output as “opt”. The sensitivity and speed of the search are inversely related and controlled by the “k-tup” variable, which specifies the size of a “word” (Pearson and Lipman). Also refers to a format for a nucleic acid or protein sequence.
- fingerprint
The pattern of bands on a gel produced by a clone when restricted by a particular enzyme, such as HindIII.
- finished sequence
High-quality, low-error DNA sequence that is free of gaps. To qualify as a finished sequence, only a single error out of every 10,000 bases (i.e., an accuracy of 99.999%) is allowed.
- FISH
Fluorescence in situ hybridization. In this technique, fluorescent molecules are used to label a DNA probe, which can then hybridize to a specific DNA sequence in a chromosome spread so that the site becomes visible through a microscope. FISH has been used to highlight the locations of genes, subchromosome regions, entire chromosomes, or specific DNA sequences. It has been used for mapping and the detection of genomic rearrangements, as well as studies on DNA replication.
- flatfile or flat file
A flat file is a data file that contains records (each corresponding to a row in a table); however, these records have no structured relationships. To interpret these files, the format properties of the file should be known. For example, a database management system may allow the user to export data to a comma-delimited file. Such a file is called a flat file because it has no inherent information about the data, and interpretation requires additional information. Files in a database management system have more complex storage structures.
- freeze
To copy changing data so as to preserve the dataset as it existed at a particular point in time. Also used to refer to the resulting set of frozen data.
- FTP
File Transfer Protocol. A method of retrieving files over a network directly to the user's computer or to his/her home directory using a set of protocols that govern how the data are to be transported.
- gap
A gap is a space introduced into an alignment to compensate for insertions and deletions in one sequence relative to another. To prevent the accumulation of too many gaps in an alignment, introduction of a gap causes the deduction of a fixed amount (the gap score) from the alignment score. Extension of the gap to encompass additional nucleotides or amino acid is also penalized in the scoring of an alignment. (See the figure for more information.)
- GB
gigabytes
- GBFF
GenBank Flat File. Refers to a format .gbff.
- GenBank
GenBank is a database of nucleotide sequences from more than 100,000 organisms. Records that are annotated with coding region features also include amino acid translations. GenBank belongs to an international collaboration of sequence databases that also includes EMBL and DDBJ. [See the GenBank chapter (Chapter 1) or the GenBank web page.]
- GeneID
GeneID is a unique identifier that is assigned to a gene record in Entrez Gene. It is an integer and is species specific. In other words, the integer assigned to dystrophin in human is different from that in any other species. For genomes that had been represented in LocusLink, the GeneID is the same as the LocusID. The GeneID is reported in RefSeq records as a 'db_xref' (e.g. /db_xref="GeneID:856646", in GenBank format).
- genetic code
The instructions in a gene that tell the cell how to make a specific protein. A, T, G, and C are the “letters” of the DNA code; they stand for the chemicals adenine, thymine, guanine, and cytosine, respectively, that make up the nucleotide bases of DNA. Each gene's code combines the four chemicals in various ways to spell out three-letter “words” that specify which amino acid is needed at every position for making a protein.
- GenomeScan
A gene identification algorithm that is used to identify exon–intron structures in genomic DNA sequence.
- genotype
The genetic identity of an individual that does not show as outward characteristics. The genotype refers to the pair of alleles for a given region of the genome that an individual carries.
- GEO
Gene Expression Omnibus. GEO is a gene expression data repository and online resource for the retrieval of gene expression data from any organism or artificial source. Many types of gene expression data from platform types, such as spotted microarray, high-density oligonucleotide array, hybridization filter, and serial analysis of gene expression (SAGE) data, are accepted, accessioned, and archived as a public dataset. [See the GEO chapter (Chpater 6) or the GEO web page.]
- GI
The GenInfo Identifier is a sequence identification number for a nucleotide sequence. If a nucleotide sequence changes in any way, a new GI number will be assigned. A separate GI number is also assigned to each protein translation within a nucleotide sequence record, and a new GI is assigned if the protein translation changes in any way. GI sequence identifiers run parallel to the new accession.version system of sequence identifiers (see the description of Version).
- GSS
Genome Survey Sequences are analogous to ESTs except that the sequences are genomic in origin, rather than cDNA (mRNA). The GSS division of GenBank contains (but is not limited to) the following types of data: random “single-pass read” genome survey sequences, cosmid/BAC/YAC end sequences, exon-trapped genomic sequences, and Alu-PCR sequences.
- heterozygosity
The probability that a diploid individual will have two different alleles at a particular genome locus. These individuals are defined as heterozygous, whereas individuals who have two identical alleles at the locus are defined as homozygous. The probability can be estimated by sampling a representative number of individuals from the population and dividing the number of heterozygotes by the total number sampled.
- HIV
Human Immunodeficiency Virus. HIV-1 is a retrovirus that is recognized as the causative agent of AIDS (Acquired Immunodeficiency Syndrome).
- HNPCC
Hereditary nonpolyposis colon cancer
- homogeneously staining region
A region of the chromosome identified cytologically by DNA staining or the FISH technique because of the presence of multiple copies of a subchromosomal region resulting from amplification.
- homologous
The term refers to similarity attributable to descent from a common ancestor. Homologous chromosomes are members of a pair of essentially identical chromosomes, each derived from one parent. They have the same or allelic genes with genetic loci arranged in the same order. Homologous chromosomes synapse during meiosis.
- HTGS
High-Throughput Genomic Sequences. The source of HTGS are large-scale genome sequencing centers; unfinished sequences are in phases 0, 1, and 2, and finished sequences are in phase 3.
- HTGS_CANCELLED
A keyword added to GenBank entries by sequencing centers to indicate that work has stopped on a clone and that the existing sequence will not be finished. Sequencing centers may stop work because the clone is redundant or for various other reasons.
- HTGS_PHASE0, HTGS_PHASE1, HTGS_PHASE2, HTGS_PHASE3
Keywords added to GenBank entries by sequencing centers to indicate the status (phase) of the sequence (see phase definitions described under draft sequence).
- HTML
Hypertext Markup Language. HTML is derived from SGML. It is a text-based mark-up language and is used to primarily display information using a web browser and to link pieces of information via hyperlinks. The tags used in an HTML document provide information only on how the content is to be displayed but do not provide information about the content they encompass.
- HUP
Hold Until Published. HUP refers to the category for data that is electronically submitted for when it should be released to the public.
- ICBN
International Code of Botanical Nomenclature
- ICD
International Classification of Diseases
- ICD-O-3
International Classification of Diseases for Oncology, 3rd edition
- ICNB
International Code of Nomenclature of Bacteria
- ICNCP
International Code of Nomenclature for Cultivated Plants
- ICTV
- ICVCN
- ICZN
- ideogram
A diagrammatic representation of the karyotype of an organism.
- IMAGE Consortium
Integrated Molecular Analysis of Genomes and their Expression. A consortium of academic groups that share high-quality, arrayed cDNA libraries and place sequence, map, and expression data of the clones in these arrays into the public domain. With the use of this information, unique clones can be rearrayed to form a “master array”, with the aim of ultimately having a representative cDNA from every gene in the genome under study. To date, human, mouse, rat, zebrafish, and Xenopus laevis genomes have been studied.
- intron
Refers to that portion of the DNA sequence that is present in the primary transcript and that is removed by splicing during RNA processing and is not included in the mature, functional mRNA, rRNA, or tRNA. Also called an intervening sequence. (See also splice sites.)
- ISAM
Indexed Sequential-Access Method. ISAM is a database access method. It allows data records in a database to be accessed either sequentially (in the order in which they were entered) or randomly (using an index). In the index, each record has a unique key that enables its rapid location. The key is the field used to reference the record.
- ISCN
International System for Human Cytogenetic Nomenclature
- ISO
- ISSN
International Standard Serial Number. The ISSN is an eight-digit number that identifies periodical publications, including electronic serials.
- karyotype
The particular chromosome complement of an individual or a related group of individuals, as defined by both the number and morphology of the chromosomes, usually in mitotic metaphase, and arranged by pairs according to the standard classification.
- LANL
- LIMS
Laboratory Information Management Systems. LIMS comprise software that helps biological and chemical laboratories handle data generation, information management, and data archiving.
- LinkOut
A registry service to create links from specific articles, journals, or biological data in Entrez to resources on external web sites. Third parties can provide a URL, resource name, brief description of their web sites, and specification of the NCBI data from which they would like to establish links. The specification can be written as a valid Boolean query to Entrez or as a list of identifiers for specific articles or sequences. Entrez PubMed users can then select which external links are visible in their searches through the NCBI Cubby service (see above). (See the LinkOut chapter or web page.)
- locus
In a genomic contect, locus refers to position on a chromosome. It may, therefore, refer to a marker, a gene, or any other landmark that can be described.
- MACAW
Multiple Alignment Construction and Analysis Workbench. MACAW is a program for locating, analyzing, and editing blocks of localized sequence similarity among multiple seqences and linking them into a composite multiple alignment.
- Map Viewer
The Map Viewer is a software component of Entrez Genomes that provides special browsing capabilities for a subset of organisms. It allows one to view and search an organism's complete genome, display chromosome maps, and zoom into progressively greater levels of detail, down to the sequence data for a region of interest. If multiple maps are available for a chromosome, it displays them aligned to each other based on shared marker and gene names and, for the sequence maps, based on a common sequence coordinate system. The organisms currently represented in the Map Viewer are listed in the Entrez Map Viewer help document, which provides general information on how to use that tool. The number and types of available maps vary by organism and are described in the “data and search tips” file provided for each organism.
- MB
megabytes
- MEDLINE
MEDLINE is NLM's database of indexed journal citations and abstracts in the fields of biomedicine and healthcare. It encompasses nearly 4,500 journals published in the United States and more than 70 other countries. (For more information, see the Fact Sheet.)
- MegaBLAST
MegaBLAST is a program for aligning sequences that differ slightly as a result of sequencing or other similar “errors”. When larger word size is used, it is up to 10 times faster than more common sequence-similarity programs. MegaBLAST is also able to efficiently handle much longer DNA sequences than the blastn program of the traditional BLAST algorithm. It uses the GREEDY algorithm for a nucleotide sequence alignment search.
- MeSH
Medical Subject Headings. MeSH refers to the controlled vocabulary of NLM used for indexing articles in PubMed. MeSH terminology provides a consistent way to retrieve information that may use different terminology for the same concepts. (See the MeSH homepage.)
- Metathesaurus
Metathesaurus is a National Cancer Institute browser containing different biomedical vocabularies, including the International Classification of Diseases for Oncology ICD-O-3.
- mFASTA
Multi-FASTA format.
- MGC
Mammalian Gene Collection. MGC is a project of the NIH to provide a complete set of full-length (open reading frame) sequences and cDNA clones of expressed genes for human and mouse.
- MGD
Mouse Genome Database. MGD contains information on mouse genetic markers, molecular segments, phenotypes, comparative mapping data, experimental mapping data, and graphical displays for genetic, physical, and cytogenetic maps.
- MGI
Mouse Genome Informatics. MGI houses a database that provides integrated access to data on the genetics, genomics, and biology of the laboratory mouse.
- microsatellite
Repetitive stretches of short sequences of DNA used as genetic markers to track inheritance in families (e.g., CC[TATATATA]CCCT). Also known as short tandem repeats (STRs).
- MIM
Mendelian Inheritance in Man. First published in 1966, Mendelian Inheritance in Man (MIM) is a genetic knowledge base that serves clinical medicine and biomedical research, including the Human Genome Project.
- minimal tiling path
An ordered list or map that defines the minimal set of overlapping clones needed to provide complete coverage of a chromosome or other extended segment of DNA (compare with tiling path).
- MMDB
Molecular Modeling Database. MMDB is a database of three-dimensional biomolecular structures derived from X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy.
- MMDB-ID
Molecular Modeling Database Accession number.
- mRNA
messenger RNA. mRNA describes the section of a genomic DNA sequence that is transcribed, and can include the 5' untranslated region (5'UTR), CDS, and 3' untranslated region (3'UTR). Successful translation of the CDS section of an mRNA results in the synthesis of a protein.
- mutation
A permanent structural alteration in DNA. In most cases, DNA changes have either no effect or cause harm, but occasionally a mutation can improve an organism's chance of surviving, and the beneficial change is passed on to the organism's descendants. Typically, mutations are more rare than polymorphisms in population samples because natural selection recognizes their lower fitness and removes them from the population.
- NCBI
- NCBI Toolkit
Contains supported software tools from the Information Engineering Branch (IEB) of the NCBI. The NCBI Toolkit describes the three components of the ToolBox: data model, data encoding, and programming libraries. Provides access to documentation for the DataModel, C Toolkit, C++ Toolkit, NCBI C Toolkit Source Browser, XML Demo Program, XML DTDs, and the FTP site.
- NCI
- NEXUS
NEXUS refers to a file format designed to contain data for processing by computer programs. NEXUS files should end with .nxs or .nex for purposes of clarity (Maddison et al., Syst Biol 46:590-621; 1997).
- NIH
- NLM
- NMR
Nuclear Magnetic Resonance. NMR is a spectroscopic technique used for the determination of protein structure.
- nr-PDB
non-redundant Protein Data Bank
- OMIM
Online Mendelian Inheritance in Man. OMIM is a directory of human genes and genetic disorders, with links to literature references, sequence records, maps, and related databases.
- ortholog
Orthology describes genes in different species that derive from a single ancestral gene in the last common ancestor of the respective species.
- orthology
Orthology describes genes in different species that derive from a common ancestor, i.e., they are direct evolutionary counterparts.
- paralog
A paralog is one of a set of homologous genes that have diverged from each other as a consequence of gene duplication. For example, the mouse α-globin and β-globin genes are paralogs. The relationship between mouse α-globin and chick β-globin is also considered paralogous.
- paralogy
Paralogy describes the relationship of homologous genes that arose by gene duplication.
- PCR
Polymerase Chain Reaction. A technique for amplifying a specific DNA segment in a complex mixture. Also present in the DNA mixture are short oligonucleotide primers to the DNA segment of interest and reagents for DNA synthesis. PCR relies on the ability of DNA to separate into its two complementary strands at high temperature (a process called denaturation) and for the two strands to anneal at an optimal lower temperature (annealing). The annealing phase is followed by a DNA synthesis step at an optimal temperature for a heat-stable DNA polymerase. After multiple rounds of denaturation, annealing, and DNA synthesis, the DNA sequence specified by the oligonucleotide primers is amplified.
- PDB
Protein Data Bank. The PDB is a database for 3D macromolecular structure data.
- Pfam
Pfam is a database housing a large collection of multiple sequence alignments and hidden Markov models covering many common protein domains.
- phenotype
The observable traits or characteristics of an organism, e.g., hair color, weight, or the presence or absence of a disease. Phenotypic traits are not necessarily genetic.
- PHRAP
A computer program that assembles raw sequence into sequence contigs (see above) and assigns to each position in the sequence an associated “quality score”, on the basis of the PHRED scores of the raw sequence reads. A PHRAP quality score of X corresponds to an error probability of approximately 10-X/10 . Thus, a PHRAP quality score of 30 corresponds to 99.9% accuracy for a base in the assembled sequence.
- PHRED
A computer program that analyses raw sequence to produce a “base call” with an associated “quality score” for each position in the sequence. A PHRED quality score of X corresponds to an error probability of approximately 10-X/10 . Thus, a PHRED quality score of 30 corresponds to 99.9% accuracy for the base call in the raw read.
- phyletic pattern
Pattern of presence–absence of a cluster of orthologs (COG) in different species.
- PHYLIP
PHYLogeny Inference Package. A package of programs for various computer platforms to infer phylogenies or evolutionary trees, freely available from the Web.
- PIR
- PMC
PubMed Central. NLM's digital archive of life sciences journal literature.
- PMID
PubMed ID number
- PNG
Portable Network Graphics. An extensible file format for the lossless, well-compressed storage of raster images (images that are composed of horizontal lines of pixels, such as those created by a computer screen). Compression of image, media, and application files is necessary to reduce the transmission time across the web. The technique of lossless compression reduces the size of the file without sacrificing any original data, and the image after expansion is exactly as it was before compression. PNG overcomes the patent issues of GIF (Graphic Interchange Format) and can replace many common uses of TIFF (Tagged Image File Format). Several features such as indexed color, grayscale, and truecolor are supported, as well as an optional alpha-channel. PNG is designed to work well in online viewing applications and is supported as an image standard by the WWW.
- poly A
A string of adenylic acid residues that are added to the 3′ end of the primary mRNA transcript. Poly(A) polymerase is the enzyme that adds the poly A tail, which is between 100 and 250 bases long.
- polymorphism
A common variation in the sequence of DNA among individuals. Genetic variations occurring in more than 1% of the population would be considered useful polymorphisms for genetic linkage analysis.
- polypeptide
Linear polymer of amino acids connected by peptide bonds. Proteins are large polypeptides, and the two terms are commonly used interchangeably.
- PRF
- private polymorphism
Variations that are only common in specific populations. Usually such populations are reproductively isolated from other, larger groups. These variations may be completely absent in other groups.
- ProtEST
A database of protein sequences from eight organisms: human (Homo sapiens), mouse (Mus musculus), rat (Rattus norvegicus), fruitfly (Drosophila melanogaster), worm (Caenorhabditis elegans), yeast (Saccharomyces cerevisiae), plant (Arabidopsis thaliana), and bacteria (Escherichia coli). (See the ProtEST web page.)
- PROW
Protein Reviews On the Web. An online resource that features PROW Guides—authoritative, short, structured reviews on proteins and protein families. The Guides provide approximately 20 standardized categories of information (abstract, biochemical function, ligands, references, etc.) for each protein.
- pseudogene
A sequence of DNA that is very similar to a normal gene but that has been altered slightly so that it is not expressed. Such genes were probably once functional but, over time, acquired one or more mutations that rendered them incapable of producing a protein product.
- PSI-BLAST
Position-Specific Iterated BLAST. PSI-BLAST (Altschul et al., J Mol Biol 215:403-410; 1990) is used for iterative protein–sequence similarity searches using a position-specific score matrix (PSSM). It is a program for searching protein databases using protein queries to find other members of the same protein family. All statistically significant alignments found by BLAST are combined into a multiple alignment, from which a PSSM is constructed. This matrix is used to search the database for additional significant alignments, and the process may be iterated until no new alignments are found.
- PSSM
Position-Specific Score Matrix. The PSSM gives the log-odds score for finding a particular matching amino acid in a target sequence.
- PubMed
A retrieval system containing citations, abstracts, and indexing terms for journal articles in the biomedical sciences. It includes literature citations supplied directly to NCBI by publishers as well as URLs to full text articles on the publishers' web sites. PubMed contains the complete contents of the MEDLINE and PREMEDLINE databases. It also contains some articles and journals considered out of scope for MEDLINE, based on either content or on a period of time when the journal was not indexed and, therefore, is a superset of MEDLINE.
- PXML
PubMed Central XML file
- QBLAST
A queuing system to BLAST that allows users to retrieve their results at their convenience and format their results multiple times with different formatting options.
- QTL
Quantitative Trait Locus. A QTL is a hypothesis that a certain region of the chromosome contains genes that contribute significantly to the expression of a complex trait. QTLs are generally identified by comparing the linkage of polymorphic molecular markers and phenotypic trait measurements. The density of the linkage map is important in the accurate and precise location of QTLs; the higher the map density, the more precise the location of the putative QTL, although there is increased likelihood that false positives will be detected. Once QTLs have been mapped to a relatively small chromosomal region, other molecular methods can be used to isolate specific genes.
- RCSB
Research Collaboratory for Structural Bioinformatics. RCSB is a nonprofit consortium that works toward the elucidation of biological, macromolecular, 3-D structures.
- Reciprocal best hits
Reciprocal best hits are proteins from different organisms that are each other's top BLAST hit, when the proteomes from those organisms are compared to each other. For example, proteins A–Z in organism 1 are compared against proteins AA–ZZ in organism 2. If protein A has a best hit to protein RR, and RR's best hit, when it is compared to all the proteins in organism 1, also turns out to protein A, then A and RR are reciprocal best hits. However, if RR's best hit is to B rather than to A, then A and RR are not reciprocal best hits.
- RefSeq
RefSeq is the NCBI database of reference sequences; a curated, non-redundant set including genomic DNA contigs, mRNAs and proteins for known genes, and entire chromosomes.
- RepeatMasker
Program that screens DNA sequences for interspersed repeats and low-complexity DNA sequences.
- RFLP
Restriction Fragment Length Polymorphism. Genetic variations at the site where a restriction enzyme cuts a piece of DNA. Such variations affect the size of the resulting fragments. These sequences can be used as markers on physical maps and linkage maps. RFLP is also pronounced “rif lip”.
- RH map
Radiation Hybrid map. A genome map in which STSs are positioned relative to one another on the basis of the frequency with which they are separated by radiation-induced breaks. The frequency is assayed by analyzing a panel of human–hamster hybrid cell lines. These hybrids are produced by irradiating human cells, which damages the cells and fragments the DNA. The dying human cells are fused with thymidine kinase negative (TK−) live hamster cells. The fused cells are grown under conditions that select against hamster cells and favor the growth of hybrid cells that have taken up the human TK gene. In the RH maps, the unit of distance is centirays (cR), denoting a 1% chance of a break occurring between two loci.
- RNA
Ribonucleic Acid. A single-stranded nucleic acid, similar to DNA, but having a ribose sugar, instead of deoxyribose, and uracil instead of thymine as one of its bases.
- RPS-BLAST
Reverse Position-Specific BLAST. A program used to identify conserved domains in a protein query sequence. It does this by comparing a query protein sequence to position-specific score matrices (PSSM)s that have been prepared from conserved domain alignments. RPS-BLAST is a “reverse” version of position-specific iterated BLAST (PSI-BLAST); however, RPS-BLAST compares a query sequence against a database of profiles prepared from ready-made alignments, whereas PSI-BLAST builds alignments starting from a single protein sequence.
- SAGE
Serial Analysis of Gene Expression. An experimental technique designed to quantitatively measure gene expression.
- Sequin
Sequin is a stand-alone software tool developed by the NCBI for submitting and updating entries to the GenBank, EMBL, or DDBJ sequence databases. It is capable of handling simple submissions that contain a single, short mRNA sequence and complex submissions containing long sequences, multiple annotations, segmented sets of DNA, or phylogenetic and population studies.
- SGD
Saccharomyces Genome Database. A database for the molecular biology and genetics of Saccharomyces cerevisceae, also known as baker's yeast.
- SGML
Standard Generalized Markup Language. The international standard for specifying the structure and content of electronic documents. SGML is used for the markup of data in a way that is self-describing. SGML is not a language but a way of defining languages that are developed along its general principles. A subset of SGML called XML is more widely used for the markup of data. HTML (Hypertext Markup Language) is based on SGML and uses some of its concepts to provide a universal markup language for the display of information and the linking of different pieces of that information.
- SKY
Spectral Karyotyping. SKY is a technique that allows for the visualization of all of an organism's chromosomes together, each labeled with a different color. This is achieved by using chromosome-specific, single-stranded DNA probes (each labeled with a different fluorophore) to hybridize or bind to the chromosomes of a cell; resulting in each chromosome being painted a different color. This technique is useful for identifying chromosome abnormalities because it is easy to spot instances where a chromosome painted in one color has a small piece of another chromosome, painted in a different color, attached to it. (Also see FISH, CGH.)
- SKYGRAM
1. A software tool to automatically convert the short-form karyotype into an image representation of a cell or clone, with each chromosome displayed in a different color, with band overlay. The program will also incorporate the number of cells for each structural abnormality, which is displayed in brackets. 2. The full ideogram or a cell or clone, with each chromosome displayed in a different color, with band overlay.
- SMART
Simple Modular Architecture Research Tool. A tool to allow automatic identification and annotation of domains in user-supplied protein sequences. For example, the SWISS-PROT database is an extensively annotated and nonredundant collection of protein sequences. SWISS-PROT annotations have been mined for SMART-derived annotations of alignments.
- SMD
Stanford Microarray Database. SMD stores raw and normalized data from microarray experiments, as well as their corresponding image files. In addition, the SMD provides interfaces for data retrieval, analysis, and visualization. Data are released to the public at the researcher's discretion or upon publication.
- SNP
Common, but minute, variations that occur in human DNA at a frequency of 1 every 1,000 bases. An SNP is a single base-pair site within the genome at which more than one of the four possible base pairs is commonly found in natural populations. Several hundred thousand SNP sites are being identified and mapped on the sequence of the genome, providing the densest possible map of genetic differences. SNP is pronounced “snip”.
- SOFT
Simple Omnibus Format in Text. SOFT is an ASCII text format that was designed to be a machine-readable representation of data retrieved from, or submitted to, the Gene Expression Omnibus (GEO). SOFT is also a line-based format, making it easy to parse, using commonly available text processing and formatting languages. (For examples of SOFT, see the guide.)
- splice sites
Refers to the location of the exon-intron junctions in a pre-mRNA (i.e., the primary transcript that must undergo additional processing to become a mature RNA for translation into a protein). Splice sites can be determined by comparing the sequence of genomic DNA with that of the cDNA sequence. In mRNA, introns (non-protein coding regions) are removed by the splicing machinery; however, exons can also be removed. Depending on which exons (or parts of exons) are removed, different proteins can be made from the same initial RNA or gene. Different proteins created in this way are “splice variants” or “alternatively spliced”.
- SSAHA
Sequence Search and Alignment by Hashing Algorithm. SSAHA is a software tool for very fast matching and alignment of DNA sequences and is used for searching databases containing large amounts (gigabases) of genome sequence. It achieves its fast search speed by converting sequence information into a “hash table” data structure, which can then be searched very rapidly for matches (Ning et al., Genome Res 11:1725-1729; 2001).
- SSLP
Simple Sequence Length Polymorphisms. SSLPs are markers based on the variation in the number of short tandem repeats in DNA.
- STS
A short DNA segment that occurs only once in the human genome, the exact location and order of bases of which are known. Because each is unique, STSs are helpful for chromosome placement of mapping and sequencing data from many different laboratories. STSs serve as landmarks on the physical map of the human genome.
- substitution matrix
A substitution matrix containing values proportional to the probability that amino acid i mutates into amino acid j for all pairs of amino acids. Such matrices are constructed by assembling a large and diverse sample of verified pairwise alignments of amino acids. If the sample is large enough to be statistically significant, the resulting matrices should reflect the true probabilities of mutations occurring through a period of evolution. (See also BLOSUM 62.)
- SWISS-PROT
SWISS-PROT is a curated protein sequence database that provides a high level of annotation (such as the description of protein function, domain structures, post-translational modifications, variants, etc.), a minimal level of redundancy, and high level of integration with other databases.
- Sybase
A trademarked family of products that include databases, development tools, integration middleware, enterprise portals, and mobile and wireless servers.
- synteny
On the same strand. The phrase “conserved synteny” refers to conserved gene order on chromosomes of different, related species.
- Tax BLAST
BLAST Taxonomy Reports page. Tax BLAST groups BLAST hits by source organism, according to information in NCBI's Taxonomy database. Species are listed in order of sequence similarity with the query sequence, the strongest match listed first.
- taxID
Taxonomy Identifier. The taxID is a stable unique identifier for each taxon (for a species, a family, an order, or any other group in the taxonomy database). The taxID is seen in the GenBank records as a “source” feature table entry; for example, /db_xref=“taxon:<9606>” is the taxID for Homo sapiens, and the line is therefore found in all recent human sequence records.
- taxid
See taxID.
- termination codon or stop codon
One of three codons that do not specify any amino acid and hence causes translation of mRNA into protein to be terminated. These codons mark the end of a protein coding sequence.
- TIGR
- tiling path
An ordered list or map that defines a set of overlapping clones that covers a chromosome or other extended segment of DNA.
- TPA
Third-Party Annotation
- TPF
Tiling Path Format. A table format used to specify the set of clones that will provide the best possible sequence coverage for a particular chromosome, the order of the clones along the chromosome, and the location of any gaps in the clone tiling path. Also used to refer to a file (Tiling Path File) in which the minimal tiling path of clones covering a chromosome is specified in Tiling Path Format or to the minimal tiling path of clones so defined.
- translation start site
The position within an mRNA at which synthesis of a protein begins. The translation start site is usually an AUG codon, but occasionally, GUG or CUG codons are used to initiate protein synthesis.
- UID
Unique Identifier
- UMLS
Unified Medical Language System. A project of the National Library of Medicine for the development and distribution of multipurpose, electronic “Knowledge Sources”, and associated lexical programs. The purpose of the UMLS is to aid the development of systems that help health professionals and researchers retrieve and integrate electronic biomedical information from a variety of sources and to make it easy for users to link disparate information systems, including computer-based patient records, bibliographic databases, factual databases, and expert systems.
- unfinished sequence
See draft sequence.
- UniGene cluster
ESTs and full-length mRNA sequences organized into clusters such that each represents a unique known or putative gene within the organism from which the sequences were obtained. UniGene clusters are annotated with mapping and expression information when possible (e.g., for human) and include cross-references to other resources. Sequence data can be downloaded by cluster through the UniGene web pages, or the complete dataset can be downloaded from the repository/UniGene directory of the FTP site.
- UniSTS
UniSTS presents a unified, non-redundant view of sequence-tagged sites (STSs). UniSTS integrates marker and mapping data from a variety of public resources. If two or more markers have different names but the same primer pair, a single STS record is presented for the primer pair, and all the marker names are shown.
- UNIX
UNIX is an operating system that was developed by Dennis Ritchie and Kenneth Thompson at Bell Labs more than 30 years ago. It allows multitasking and multiuser capabilities and offers portability with other operating systems. It comes with hundreds of programs that are of two types: integral utilites, such as the command line interpreter; and tools such as email, which are not necessary for the operation of UNIX but provide additional capabilities to the user. It is functionally organized at three levels: the kernel, which schedules tasks and manages storage; the shell, which connects and interprets user's commands, calls programs from memory, and executes them; and tools and applications, which offer additional functionality to the operating system, such as word processing and business applications. UNIX® was registered by Bell Laboratories as a trademark for computer operating systems. Today, this mark is owned by The Open Group.
- URL
Uniform Resource Locator. The address of a resource on the Internet. URL syntax is in the form of protocol://host/localinfo, where “protocol” specifies the means of fetching the object (such as HTTP, used by WWW browsers and servers to exchange information, or FTP), “host” specifies the remote location where the object resides, and “localinfo” is a string (often a file name) passed to the protocol handler at the remote location. Also called Uniform Resource Identifier (URI).
- UTF-8
UCS (Universal Character Set) Transformation Format. An AscII-preserving encoding method for Unicode (a standard to provide a unique number for every character irrespective of the platform, program, or language).
- UTR
Untranslated Region. The 3′ UTR is that portion of an mRNA from the position of the last codon that is used in translation to the 3′ end. The 5′ UTR is that portion of an mRNA from the 5′ end to the position of the first codon used in translation.
- VAST
Vector Alignment Search Tool. A computer algorithm used to identify similar protein 3D structures.
- weight
An assignment of importance to a term in a search query. If a term in a search query is found to match a word in a document, that word is given a “weight”. The exact weight of the word will depend on the emphasis given to the word by the author or its position in the document. For example, a word that occurs in a chapter title will have a higher weight than the same word if it occurs in the body of the chapter. Similarly, words that occur in data collections are also assigned weights, depending on how frequently the terms occur in the collection.
- WGS sequence
Whole Genome Shotgun sequence. In this semi-automated sequencing technique, high-molecular-weight DNA is sheared into random fragments, size selected (usually 2, 10, 50, and 150 kb), and cloned into an appropriate vector. The clones are then sequenced from both ends. The two ends of the same clone are referred to as mate pairs. The distance between two mate pairs can be inferred if the library size is known and has a narrow window of deviation. The sequences are aligned using sequence assembly software. Proponents of this approach argue that it is possible to sequence the whole genome at once using large arrays of sequencers, which makes the whole process much more efficient than the traditional approaches.
- WHO
- WWW
World Wide Web. A consortium (W3C) that develops technologies such specifications, guidelines, software, and tools for the internet.
- XML
Extensible Markup Language. XML describes a class of data objects called XML documents and partially describes the behavior of computer programs that process them. XML is a subset of SGML, and XML documents are conforming SGML documents. XML documents are made up of storage units called entities, which contain either parsed or unparsed data. Parsed data is made up of characters (a unit of text), some of which form character data, and some of which form markup. Markup includes tags that provide information about the data, i.e., a description of the structure and content of the document. Character data comprises all the text that is not markup. XML provides a mechanism to impose constraints on the storage layout and logical structure.
- XSL
Extensible Stylesheet Language. XSL is used for the transformation of XML-based data into HTML or other presentation formats, for display in a web browser. This is a two-part process. First, the structure of the input XML tree must be transformed into a new tree (e.g., HTML), allowing reordering of the elements, addition of text, and calculations—all without modification to the source document. This process is described by XSLT. Second, XSL-FO (XSL Formatting Objects, an XML vocabulary for formatting) is used for formatting the output, defining areas of the display page and their properties. In this way, the source XML document can be maintained from the perspective of “pure content” and can be separated from the presentation. An XML document can be delivered in different formats to different target audiences by simply switching style sheets.
- XSLT
Extensible Stylesheet Language: Transformations. XSLT is a language for transforming the structure of an XML document. XSLT is designed for use as part of XSL, the stylesheet language for XML. A transformation expressed in XSLT describes a sequence of template rules for transforming a source tree into a result tree; elements from the source tree can be filtered and reordered, and a different structure can be added. A template rule has two parts: a pattern that is matched against nodes in the source tree; and a template that can be instantiated to form part of the result tree. This makes XSLT a declarative language because it is possible to specify what output should be produced when specific patterns occur in the input, which distinguishes it from procedural programming languages, where it is necessary to specify what tasks have to be performed in what order. XSLT makes use of the expression language defined by XPath (a language for addressing the parts of an XML document) for selecting elements for processing, for conditional processing, and for generating text.
- YAC
Yeast Artificial Chromosome. Extremely large segments of DNA from another species spliced into the DNA of yeast. YACs are used to clone up to one million bases of foreign DNA into a host cell, where the DNA is propagated along with the other chromosomes of the yeast cell.
- ZFIN
Zebrafish Information Network. ZFIN is a database for the zebrafish model organism that holds information on wild-type stocks, mutants, genes, gene expression data, and map markers.