Clinical Description
VLDLR cerebellar hypoplasia is a congenital non-progressive disorder characterized by cerebellar ataxia and intellectual disability.
To date, more than 50 individuals have been identified with a pathogenic variant in VLDLR [Boycott et al 2005, Glass et al 2005, Moheb et al 2008, Ozcelik et al 2008, Türkmen et al 2008, Boycott et al 2009, Kolb et al 2010, Ali et al 2012, Azmanov et al 2013, Kruer et al 2013, Schlotawa et al 2013, Sonmez et al 2013, Giorgio et al 2016, Micalizzi et al 2016, Valence et al 2016, Wilker et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Features of VLDLR Cerebellar Hypoplasia
View in own window
| Feature | Number (%) of Persons w/Feature | Comment |
|---|
| Cerebellar hypoplasia | 44/44 (100%) | |
| Pontine hypoplasia | 36/44 (81.8%) | |
| Simplified cortical gyration | 43/44 (97.7%) | |
| Cerebellar ataxia | 53/53 (100%) | Predominantly truncal; peripheral ataxia reported in some |
| Hypotonia | 28/37 (75.7%) | |
| Dysarthria | 34/42 (81.0%) | |
| Nystagmus | 11/47 (23.4%) | |
| Strabismus | 40/51 (78.4%) | |
| Cognitive impairment | 53/53 (100%) | Moderate to profound |
| Developmental delay | 53/53 (100%) | |
| Delayed ambulation | 53/53 (100%) | Independent ambulation (if achieved) often in mid-childhood. |
| Epilepsy | 7/53 (13.2%) | |
| Brisk reflexes | 31/40 (77.5%) | |
| Microcephaly | 8/38 (21.1%) | Head circumference -2SD to -4SD |
| Dysmorphism | 1/53 (1.89%) | May not be related to VLDLR-CH |
| Short stature | 19/42 (45.2%) | |
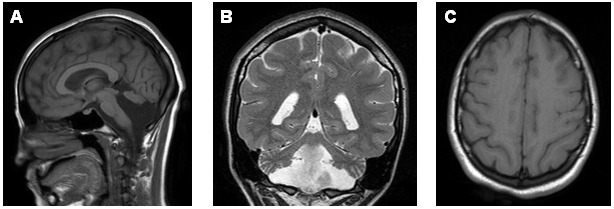
Brain MRI. All affected individuals demonstrate hypoplasia of the inferior portion of the cerebellar vermis and hemispheres. In addition, the majority of affected individuals show a simplified gyration of the cerebral hemispheres with minimally thickened but uniform cortex, lack of clear anteroposterior gradient, and small brain stem (particularly the pons). Some individuals are described as demonstrating neuroimaging features of pontocerebellar hypoplasia.
Cerebellar ataxia. All affected individuals demonstrate significant truncal ataxia. Children either learn to walk very late (often after age 6 years) or never achieve independent ambulation. For those able to ambulate independently, gait is wide based; affected individuals are not able to perform a tandem gait. Affected individuals from Turkey demonstrate quadrupedal locomotion in which the palms of the hands touch the ground and the elbows, back, and knees are straight [Ozcelik et al 2008, Türkmen et al 2009], an interesting behavioral adaptation which likely depends on the presence of special environmental influences during child development [Herz et al 2008, Türkmen et al 2009]. Limb ataxia is present in most individuals but is not severe.
Intellectual disability. All reported affected individuals have intellectual disability, ranging from moderate to profound. Most individuals can follow simple commands. Some can communicate verbally using short phrases or sentences. Adults are unable to live independently.
Dysarthria. Those who are able to communicate verbally demonstrate dysarthria.
Strabismus. The majority of individuals have strabismus.
Other
Nystagmus is reported in some individuals and is described as gaze evoked.
Epileptic seizures were reported in 40% of the affected individuals from the Hutterite population [
Glass et al 2005], and appear to be less frequent in non-Hutterite individuals. The seizures tend to be generalized.
Deep tendon reflexes in the lower extremities tend to be brisk.
Microcephaly (2-4 SD below the mean) has been reported in a few affected individuals.
Short stature (height just below the 2nd centile) is a feature in a few affected individuals.
Life span. There has been no formal study of life span in this disorder, but experience from the Hutterite population suggests that life span is not significantly reduced.
Prevalence
The actual frequency of VLDLR-CH is unknown.
More than 25 individuals with VLDLR-CH from the Hutterite population in Canada and the US have been followed for many years. This condition is present in all three Hutterite leuts (branches) (i.e., Schmiedeleut, Lehrerleut, and Dariusleut).
The estimated carrier frequency in the Hutterite population is one in 15 [Glass et al 2005].