Summary
Clinical characteristics.
Spinocerebellar ataxia type 17 (SCA17) is characterized by ataxia, dementia, and involuntary movements, including chorea and dystonia. Psychiatric symptoms, pyramidal signs, and rigidity are common. The age of onset ranges from three to 55 years. Individuals with full-penetrance alleles develop neurologic and/or psychiatric symptoms by age 50 years. Ataxia and psychiatric abnormalities are frequently the initial findings, followed by involuntary movement, parkinsonism, dementia, and pyramidal signs. Brain MRI shows variable atrophy of the cerebrum, brain stem, and cerebellum. The clinical features correlate with the length of the polyglutamine expansion but are not absolutely predictive of the clinical course.
Diagnosis/testing.
The diagnosis of SCA17 is established in a proband by identification of an abnormal CAG/CAA repeat expansion in TBP. Affected individuals usually have more than 41 repeats. The CAA and CAG codons both encode glutamine residues resulting in a pathogenic polyglutamine expansion.
Management.
Treatment of manifestations: Psychotropic medications for psychiatric issues, anti-seizure medication for seizures (ASM); botulinum toxin injections for dystonia; adaptation of the environment to accommodate dementia.
Prevention of secondary complications: Side effects of psychotropic medications and ASMs may require total or intermittent discontinuation of the treatment or reduction in dose.
Surveillance: Annual or semiannual evaluation by a neurologist or more frequently if symptoms are progressing rapidly.
Agents/circumstances to avoid: Sedative/hypnotic agents, such as ethanol or certain medications, may exacerbate incoordination.
Genetic counseling.
SCA17 is inherited in an autosomal dominant manner. Offspring of affected individuals are at a 50% risk of inheriting the expanded TBP allele. The age of onset, severity, specific symptoms, and progression of the disease are variable and cannot be precisely predicted by family history or size of expansion. Prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible if the diagnosis has been established in an affected family member by molecular genetic testing.
Diagnosis
Suggestive Findings
Spinocerebellar ataxia type 17 (SCA17) should be suspected in individuals with the following:
Ataxia
Dementia
Involuntary movements – e.g., chorea and dystonia (blepharospasm, torticollis, writer's cramp, foot dystonia)
Psychiatric symptoms
Establishing the Diagnosis
The diagnosis of SCA17 is established in a proband by identification of a heterozygous pathogenic variant of CAG (and sometimes CAA) repeats in TBP by molecular genetic testing (see Table 1). Because both codons CAA and CAG encode glutamine residues, the resulting proteins will have variable tracts of glutamine residues.
Allele sizes. The structure of the repeat sequence in a normal, stably transmitted allele is variable but typically consists of series of CAG repeats interrupted by CAA repeats – e.g., (CAG)3 (CAA)3 (CAG)9 CAA CAG CAA (CAG)16 CAA CAG. Allele size (sometimes expressed as length or number of repeats) is determined by counting all triplet repeats; the total number of CAG/CAA repeats in the example above would be 36, which would translate to 36 contiguous glutamine residues in the protein.
CAA CAG CAA interruption. The CAA CAG CAA interruption between (CAG)x and (CAG)y is present in all expanded alleles that are stably transmitted (i.e., the allele size is unchanged during meiosis).
The CAA CAG CAA interruption between (CAG)x and (CAG)y was absent in two families with allele size instability (i.e., change in allele size) during transmission [Zühlke et al 2001, Maltecca et al 2003]. Thus, loss of this interruption may be a prerequisite of instability in SCA17 as in other disorders caused by repeat expansions [Maltecca et al 2003, Zühlke et al 2003b, Zühlke et al 2005].
Molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
Single-gene testing. Targeted analysis for a
heterozygous CAG/CAA repeat number in
TBP should be performed first.
A multigene panel that includes
TBP CAG/CAA repeat analysis and other genes of interest (see
Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Table 1.
Molecular Genetic Testing Used in Spinocerebellar Ataxia Type 17
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
TBP
| Targeted analysis for CAG/CAA repeat expansion 3 | 100% |
- 1.
- 2.
- 3.
PCR amplification will likely detect CAG/CAA repeat expansions of 66 or fewer.
Clinical Characteristics
Clinical Description
Spinocerebellar ataxia type 17 (SCA17) is characterized by ataxia (95%), dementia (~90%), and involuntary movements (~70%), including chorea and dystonia (blepharospasm, torticollis, writer's cramp, foot dystonia) [Cellini et al 2004, Toyoshima et al 2004]. Psychiatric symptoms, pyramidal signs, and rigidity are common.
Onset ranges from age three to 75 years (mean: 34.6 years) [Stevanin & Brice 2008]. All individuals with full-penetrance alleles develop neurologic and/or psychiatric symptoms by age 50 years [Koide et al 1999; Fujigasaki et al 2001; Nakamura et al 2001; Zühlke et al 2001; Silveira et al 2002; Maltecca et al 2003; Stevanin et al 2003; Zühlke et al 2003b; Bauer et al 2004; Hagenah et al 2004; Oda et al 2004; Toyoshima & Takahashi 2018; Toyoshima, personal observation].
Although the disease course is variable, ataxia and psychiatric abnormalities are frequently the initial findings, followed by involuntary movement, parkinsonism, dementia, and pyramidal signs.
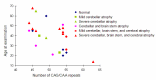
Brain MRI shows variable atrophy of the cerebrum, brain stem, and cerebellum (). Most people present with cerebellar atrophy. The age of the individual and the length of CAG/CAA repeat influence the degree of atrophy. For example, in older individuals – even those with a small full-penetrance allele – severe atrophy is present on brain MRI. High-intensity T2-weighted images and selective atrophy on caudate nucleus are not observed. Some correlation of region of brain atrophy with clinical characteristics is seen [Lasek et al 2006].
Number of CAG/CAA repeats versus age of individuals with SCA17
Neuropathology. The brain shows atrophy of the striatum (more apparent in the caudate nucleus) and cerebellum. Histologically, neuronal loss is observed in the striatum and Purkinje cell layer. Loss of cerebral cortical neurons is seen in some individuals.
Immunohistochemistry for the expanded polyglutamine (polyQ) tracts shows diffuse labeling of the neuronal nucleoplasm.
Note: Intranuclear inclusions are a much less common finding than diffuse labeling. No labeling is detectable in the cytoplasm or in the neuropil. Glial cell involvement is occasionally seen.
In individuals who are homozygous for an expanded allele in the full-penetrance range, nuclear polyQ pathology involves other CNS regions including the cerebral cortex, thalamus, and brain stem [Toyoshima et al 2004]. The abundant nuclear accumulation of polyQ in the cerebral cortices and subcortical nuclei (e.g., dorsomedian thalamic nucleus) are possibly associated with the prominent cognitive and behavioral decline in affected individuals.
Genotype-Phenotype Correlations
Heterozygotes
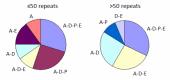
Clinical features. The length of the CAG/CAA repeat in TBP correlates with the clinical features based on data available from 52 individuals (50 from the literature and 2 unreported) (Table 2, ). As the information reported in the literature was incomplete, the frequencies listed for symptom occurrence may be underestimated [Koide et al 1999, Fujigasaki et al 2001, Nakamura et al 2001, Zühlke et al 2001, Silveira et al 2002, Maltecca et al 2003, Rolfs et al 2003, Stevanin et al 2003, Zühlke et al 2003a, Bauer et al 2004, Hagenah et al 2004, Oda et al 2004]. Of note is the high proportion of individuals with psychiatric symptoms and chorea.
The clinical features in SCA17 depend on the length of CAG/CAA repeats. The clinical features reported in affected individuals are denoted by letter. For example, A-E denotes affected individuals who have ataxia with parkinsonism. A = ataxia
Table 2.
Frequency of Clinical Features in Spinocerebellar Ataxia Type 17 Correlated with TBP Repeat Size
View in own window
CAG/CAA
Repeat Size
of Allele | Ataxia | Dementia /
Psychiatric
Symptoms | Increased
DTRs | Dystonia | Parkinsonism | Chorea |
|---|
| 41-49 | 85% | 85% | 50% | 6% | 32% | 35% |
| ≥50 | 96% | 88% | 56% | 56% | 48% | 16% |
DTRs = deep tendon reflexes
Penetrance
The penetrance of alleles of 41-44 repeats is estimated at 50% and the penetrance of alleles of 45-48 repeats is estimated at greater than 80% [Toyoshima et al 2004].
Four individuals with 42 CAG/CAA repeats developed a relatively benign
phenotype consisting of mild gait ataxia, dysarthria, and dysdiadochokinesia [
Nolte et al 2010].
An individual with 43 CAG/CAA repeats developed ataxia with dementia at age 52 years [
Silveira et al 2002]; six individuals diagnosed with parkinsonism were found to have 43 CAG/CAA repeats [
Kim et al 2009]. An individual with 43 repeats developed severe dementia [
Nielsen et al 2012].
An individual with 46 CAG/CAA repeats developed symptoms at age 75 years, the latest onset observed to date [
Wu et al 2005].
Age of onset. The correlation between the size of the CAG/CAA repeat and the age of onset in SCA17 () is not as strong as in other disorders (SCA1, SCA2, SCA3, SCA6, SCA7, Huntington disease, DRPLA, SBMA]) caused by expansion of a polyglutamine tract [Rolfs et al 2003, Toyoshima et al 2004,Toyoshima & Takahashi 2018].
Correlation between age at onset and length of CAG/CAA repeat in individuals with SCA17 For references, see Penetrance, Age of onset.
Anticipation
Instability of the TBP CAG repeat in germline transmission is not clear in SCA17 [Fujigasaki et al 2001, Nakamura et al 2001, Shatunov et al 2004]. CAG repeats in TBP have two distinct configurations, which are differentiated by the absence or presence of CAA trinucleotide repeat interruptions. The basic structure of the allele is (CAG)3 (CAA)3 (CAG)x CAA CAG CAA (CAG)y CAA CAG. If the basic structure is broken (i.e., CAA repeat interruptions are absent), repeat stability may be reduced. In German and Italian families, an absence of CAA interruptions resulting in longer pure tracts of CAG repeats was detected. It is of note that intergenerational instability and anticipation were documented in these families [Zühlke et al 2001, Maltecca et al 2003]. It has been proposed that CAA interruptions may serve as a limiting element for further expansion of CAG repeats in TBP [Gao et al 2008].
The phenomenon termed anticipation, a trend toward an earlier age at onset and more severe disease manifestations in offspring of an affected individual, is infrequently documented in families with SCA17. In addition, because of low penetrance of the intermediate alleles (41-48 repeats), the age of onset, severity, specific symptoms, and progression of the disease are variable and cannot be predicted by family history or size of expansion.
Prevalence
Fewer than 100 families with SCA17 have been reported.
The prevalence of SCA17 in the Japanese population is estimated at 0.47:1,000,000. SCA17 accounts for approximately 0.3% of autosomal dominant SCA [Maruyama et al 2002].
The minimum prevalence of SCA17 in northeast England is 0.16:100,000 [Craig et al 2005].
In a study of the Yugoslav population, none of the 115 individuals with autosomal dominant cerebellar ataxia or simplex cases of adult-onset ataxia had SCA17 [Alendar et al 2004].
The prevalence of SCA17 may be underestimated because some individuals with SCA17 have a phenotype similar to that of Huntington disease.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with spinocerebellar ataxia type 17 (SCA17), the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Neuropsychological testing to evaluate for dementia and/or psychiatric disturbance
Brain MRI to evaluate areas and degree of atrophy
Neurology consultation, if not completed prior to initial diagnosis
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with Spinocerebellar Ataxia Type 17
View in own window
| Manifestation/Concern | Treatment |
|---|
| Psychiatric symptoms | Psychotropic medications |
| Seizures | Anti-seizure medication(ASM) |
| Dystonia | Local injections of botulinum toxin |
| Dementia | Adaptation of environment |
Prevention of Secondary Complications
The side effects of psychotropic medications and ASMs (e.g., depression, sedation, nausea, restlessness, headache, neutropenia, and tardive dyskinesia) can be major secondary complications in persons with SCA17. For some individuals, the side effects of certain therapeutics may be worse than the symptoms of the disease; such individuals may benefit from total or intermittent discontinuation of the treatment or reduction in dose.
Surveillance
Affected individuals should be followed annually or semiannually by a neurologist or more frequently if symptoms are progressing rapidly, as may happen in the advanced stages [Toyoshima et al 2004].
Agents/Circumstances to Avoid
Agents with sedative/hypnotic properties, such as ethanol or certain medications, may markedly increase incoordination.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Spinocerebellar ataxia type 17 (SCA17) is inherited in an autosomal dominant manner.
Note: A novel pathomechanism, referred to as digenic TBP/STUB1-related SCA17 or SCA17-DI, is suggested by individuals with a phenotype consistent with SCA17 who are double heterozygotes for a TBP allele with 41 to 46 CAG/CAA repeats (i.e., an allele in the reduced-penetrance range) and a STUB1 pathogenic variant [Magri et al 2022, Reis et al 2022]. SCA17-DI is not discussed further in this section.
Risk to Family Members
Parents of a proband *
* Based on the family history of the 59 reported families with affected individuals [Toyoshima & Takahashi 2018]
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If one of the parents of the
proband has an expanded
TBP allele, the risk to the sibs of inheriting the expanded CAG/CAA allele is 50%. The age of onset, severity, specific symptoms, and progression of the disease are variable and cannot be precisely predicted by family history or size of expansion.
If the parents have not been tested for the expanded
TBP allele but are clinically unaffected, sibs are still presumed to be at increased risk for SCA17 because of the possibility of reduced
penetrance in a parent or the theoretic possibilities of parental
germline mosaicism or expansion from a mutable normal allele in the parent.
Offspring of a proband
Each child of an individual with SCA17 has a 50% chance of inheriting the expanded
TBP allele.
The age of onset, severity, specific symptoms, and progression of the disease are variable and cannot be precisely predicted by family history or size of expansion.
Compared with other SCA subtypes caused by expanded trinucleotide repeats,
anticipation is rare in SCA17 because CAA interruptions within the
TBP CAG repeat configuration stabilize the repeat in
germline transmission (see
Anticipation). However, if the
proband has a mild
phenotype and a short number of repeats (41-43), examination of the CAG repeat configuration to determine if CAA repeat interruptions are present can be useful in assessing the likelihood of severe anticipation in offspring.
Other family members. The risk to other family members depends on the genetic status of the proband's parents: if a parent is affected and/or is known to have an expanded TBP allele, the parent's family members are at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once a diagnosis of SCA17 has been established by molecular genetic testing in an affected family member, prenatal and preimplantation genetic testing for SCA17 are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Spinocerebellar Ataxia Type 17: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
TATA-box-binding protein (TBP) is an important general transcription initiation factor and is the DNA-binding subunit of RNA polymerase II transcription factor D, the multi-subunit complex crucial for the expression of most genes. TBP has a long tract of glutamines in the N-terminus. This region is thought to modulate the DNA binding activity of the C terminus, which affects the rate of transcription complex formation and initiation of transcription.
Mechanism of disease causation
Unknown, but the contiguous tract of polygutamine encoded by the pathogenic expanded
TBP allele is generally considered to confer a gain of function.
Because TBP is a fundamental
transcription factor expressed ubiquitously in all organs, including the CNS, the question of whether loss of TBP function plays a role in the pathogenesis of spinocerebellar ataxia Type 17 (SCA17) remains to be addressed. In a homozygote, however, no abnormality was observed in growth, and pathologic examination showed no specific changes in the visceral organs [
Toyoshima et al 2004]. Taking into consideration the ubiquitous presence of TBP, the selective neuronal degeneration suggests no significant loss of protein function in individuals with SCA17.
TBP-specific laboratory considerations
The trinucleotide CAG repeats, sometimes interrupted by CAA repeats, are in
TBP exon 3. Both CAG and CAA trinucleotide repeats encode the amino acid glutamine resulting in a contiguous tract of polyglutamine at the protein level.